Part 1: History and Significance of the accepted Gold Standard in DNA Sequencing
This is Part One of a six-part series on Sanger sequencing and Capillary Electrophoresis, starting with the history and significance of the accepted gold standard in DNA sequencing. CE sequencing applications will follow.
In 1951, Fred Sanger first determined the sequence of two proteins (the bovine insulin A and B forms), showing that they were distinct molecules, and for this he was awarded a Nobel Prize in Chemistry in 1958. In 1965, he lost the race to be the first to sequence a tRNA molecule. In 1977, he introduced the “di-deoxy” chain-termination method, which earned him a second Nobel Prize in Chemistry in 1980 (which he shared with Paul Berg and Walter Gilbert, of the Maxam-Gilbert chemical-based method of sequencing ). ‘Sanger method’ became the standard method for sequencing for its ease-of-use; in particular it did not require the use of hazardous chemicals as it was a biochemical method mimicking natural DNA synthesis.
By utilizing a mixture of natural nucleotides (deoxy-nucleotide triphosphates or dNTPs for short) with modified ones (dideoxy-nucleotide triphosphates or ddNTPs), in the normal course of DNA polymerization, a given molecule being extended will randomly add a dideoxynucleotide. Without a 3’-hydroxy group to continue polymerization (thus the name “di-deoxynucleotide”), that molecule will stop growing, and after the reaction is finished, there is a mixture of products of differing sizes.
The invention of PCR in 1985 was rapidly included into the Sanger sequencing process (so-called ‘cycle-sequencing’), and the availability of 35S-labeled dNTPs made the process relatively safer than the high gamma radiation-emitting 32P-end-labeled primers.
A graphical video of the sequencing process is available to further illustrate this.
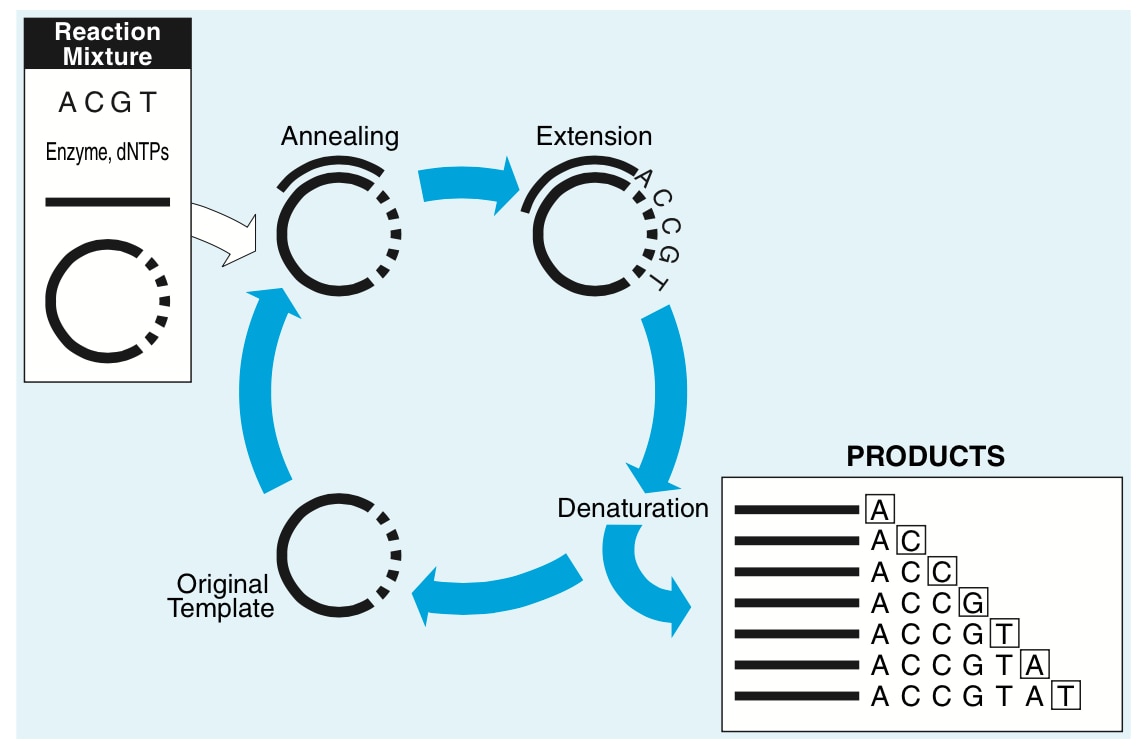
When this technique was first introduced in the late 1970’s, radioactive end-labeled primers were used and each reaction separated into four distinct reaction tubes, one for each didoxynucleotide. Thus, the tube labeled ‘A’ would have a mix of all four dNTP’s (the normal chemical nucleotides for DNA chain elongation) and ddATP. Each reaction tube would then be separated individually on large acrylamide slab electrophoresis gels (these gels were large, on the order of about 30cm x 50cm; and after drying the gel and autoradiography to X-ray film the DNA ladder could be read.
Of course this process was manual, with a fluorescent light-box to read the autoradiogram, a ruler, and (hopefully) an assistant to enter the four letters into the computer one base at a time. G. G. A. A. T. T. C. C. A… It wasn’t unusual to have to go back a second time to double-check the accuracy, and from setup of the sequencing reaction to pouring the gel and polymerizing it to running the gel to drying the gel and then to reading the gel would take about a day and a half.
A major shift in sequencing technology
Fast-forward over twenty years, and the radionucleotides have been replaced with dye-termination nucleotides. (Thermo Fisher Scientific has currently four different terminator chemistries, including the popular BigDye® Terminators v3.1.) In addition, instead of x-ray film, heavy x-ray film cassettes (with the requisite intensifying screen necessitating placing the film plus cassette in a -80C freezer overnight) and darkroom film manipulation (along with maintaining the film developer, yet another unwelcome laboratory chore), an automated detection system uses a scanning laser and a photomultiplier tube with advanced optics to read the fluorescent dye, for direct automated entry of DNA base sequence data.
While this major shift away from radioactivity and x-ray film to fluorescence and optical detection occurred with the introduction of the Applied Biosystems® 370A DNA Sequencer in 1986, the next major advance occurred when the ABI® Prism 310 genetic analyzer was introduced in 1995. The ABI® Prism 310 tackled the other rather unpleasant aspect of DNA sequencing at that time: pouring gels. These electrophoresis gel glass plates need fastidious handling, as a speck of dust can easily form an air bubble as the glass sheets between which the acrylamide is polymerized is typically 0.4mm wide. In addition, fresh acrylamide solutions are needed for optimum performance, which necessitated handling dry acrylamide, a suspected carcinogen.
Sequencing technology mirrors genomics
Only a few years after the introduction of the ABI® Prism 310, the 96-capillary AB 3700 became the workhorse of the Human Genome Project. This was a 100-fold improvement in only 5 years, a remarkable testament to the internal development teams to develop the biochemistry, polymer chemistry, and mechanical/optical engineering. In an interview (still available as a PDF here) Dr. Elaine Mardis (then co-director of the Washington University Genome Sequencing Center) said of the AB®3700: “I don’t think the human genome, wherever you look at it being done, could have been done without these instruments.” The importance of this sequencing technology to the E. coli genome, the Saccharomyces cerevisiae genome, the Caenorhabditis elegans genome, the Drosophila melanogaster genome, and the Mus musculus genome leading directly to the Homo Sapiens genome cannot be overstated, as the accelerating pace of science rose in tandem with the accelerating pace of technical development of the sequencing technology.
After these decades of technical improvement, Sanger sequencing by capillary electrophoresis has a justified claim of being the gold standard for sequence quality. Incremental improvements from protein engineering of the DNA polymerase used for sequencing (modified nucleotides with inorganic fluorescent dyes are relatively large, requiring a fair amount of engineering of the nucleotide ‘pocket’), chemical improvements of the acrylamide and other components of the POP polymer separation matrix, optical improvements to sense subtle changes in fluorescence coupled with newer dye-chemistry, and finally software improvements in base-calling and error-modeling helped enable Sanger sequencing by capillary electrophoresis to provide highly accurate results in very high accuracy.
Our current system, the Applied Biosystems™ 3500 Series Genetic Analyzer, includes a wide array of technology that enables this system to be simple to use. Everything from a solid-state long-life laser, radio-frequency identification tags (RFID) to track consumables usage and lot number information, pre-packaged polymer pouches, and data collection software are all designed for ease-of-use.
A word on accuracy
With so much improvement over time, and so many years which the system has been developed, the error model for Sanger sequencing is without equal. Quantified by a program called Phred and defined as Q = – 10 log10 P where Q is the quality score, and P is the error of probability. For example with a 10% error, the Phred quality score is 10, and the accuracy is therefore 90%. At a Phred quality score of 20, the error is 1%, and the accuracy is 99%; at 30, the error is 0.1%, and the accuracy is 99.9%.
The overall accuracy of Sanger sequencing is accepted to be on the order of 99.95%, or an overall error of 0.05%. With read-lengths of 800 to 1000 base-pairs, these two aspects of Sanger sequencing (accuracy and read-length) make it the gold standard for sequencing.
If you want to learn more, here’s a great reference resource called “DNA Sequencing by Capillary Electrophoresis – Applied Biosystems Chemistry Guide”. It is very comprehensive, and as a PDF it doesn’t take a lot of room on your desk.
References: A history of innovation in genetic science. Applied Biosystems (PDF)
Check out the whole Series:
Sanger Sequencing by CE 1: Foundations
Sanger Sequencing using CE 2: Fragment Analysis
Fragment Analysis using CE 3: Designing a 27-plex PCR
Sanger Sequencing by CE 4: Bioinformatics
Leave a Reply