Alan Livingston and Karen Jones, EMEA Market Development, Thermo Fisher Scientific
Spinal Muscular Atrophy (SMA) is the second most common fatal autosomal recessive disorder with a carrier frequency of 1 in 54-57 in all populations and incidence of ~1 in 6,000 to 10,000 live births. It is a serious condition that gets worse as over time and is caused by mutations in both copies of the SMN1 gene located on chromosome 5.
There are four types of SMA which range from severe (onset in early infancy) to mild (onset in young adulthood), respectively. Babies with type 1 rarely live beyond the first few years following birth. Overall, approximately 60% of individuals with SMA are severely affected and die in early childhood.
Highly homologous genes complicate genetic analysis
The majority of mutations responsible for SMA are either deletions or gene conversions. The complication in identifying carriers of SMN1 mutations is that humans have two nearly identical copies of SMN1 and SMN2 located on Chromosome 5; the major difference between SMN1/SMN2 is found at the beginning of exon 7. There is a single nucleotide difference – C for SMN1 and T for SMN2. Additionally SMN1 mRNA includes exon 7 whereas the SMN2 mRNA generally excludes exon 7 (fig 1). The presence of exon 7 is critical for the production of fully functional and stable SMN protein.
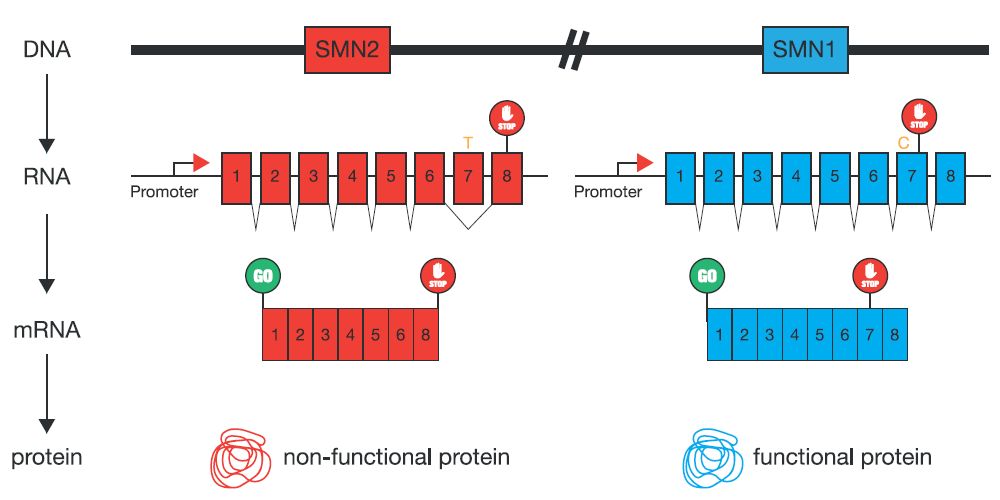
Figure 1 Mutations in SMN1 contribute to SMA
About 5% of 5q-SMA patients have a deletion or gene conversion mutation on one chromosome and a point mutation on the other chromosome. An individual with this combination of mutations (point mutation with either a deletion or conversion mutation) will not be diagnosed as having SMA using the SMA diagnostic test as only one copy of the SMN1 gene is gone. Rather, this person will look like a carrier using the quantitative carrier test, even though they are symptomatic for SMA.
In both scenarios, deletion and gene conversion, SMA patients are missing SMN1 exon 7, referred to as homozygous absence of SMN1 exon 7. Although patients with SMA have mutations in SMN1, they always carry at least one normal copy of SMN2. The homozygous loss of both genes has not been reported and is assumed lethal.
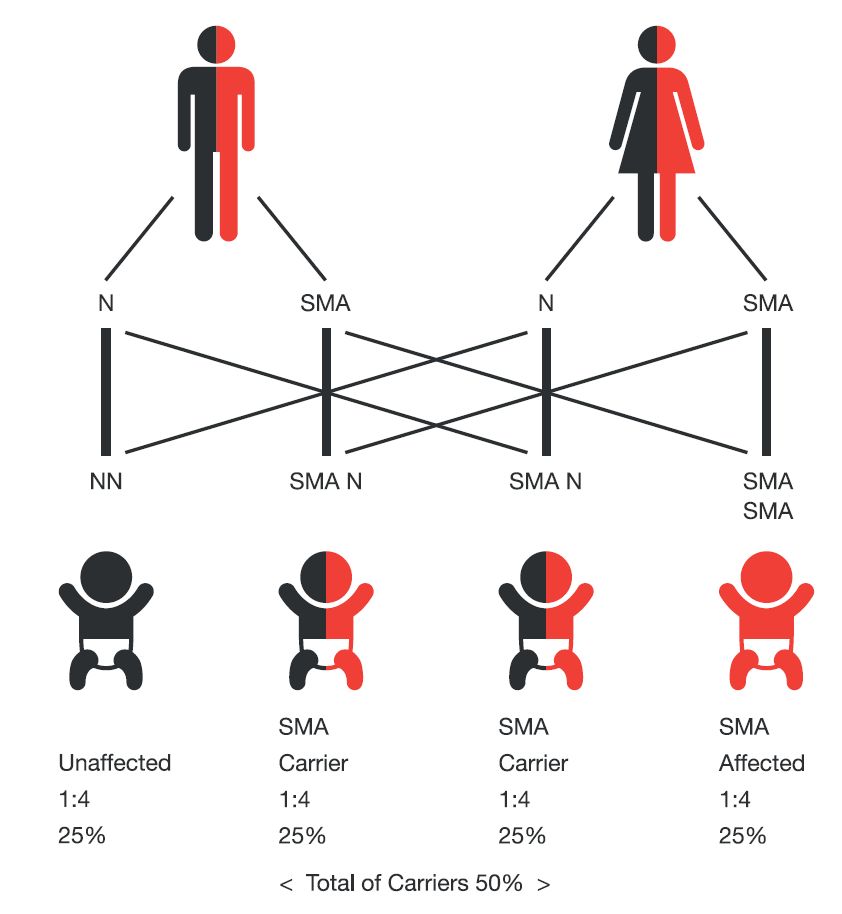
Figure 2 Transmission of the SMN1 gene mutation in SMA
Some individuals have been identified with two copies of SMN1 on one chromosome and a SMN1 deletion on the other chromosome (2 + 0 SMA carrier status), and are termed as silent carriers.
Having a child affected by SMA occurs in a pregnancy between two SMA carriers or between a SMA carrier and a person living with SMA. Two carrier parents can produce children who would be affected, carriers, or non-carriers. In these families, each pregnancy has a:
- 25% chance of producing a child who would be affected with SMA
- 50% chance of producing a child who would be a SMA carrier
- 25% chance of producing a child who would not have SMA and would not be a SMA carrier
Current testing regimes
These include: a blood test for creatine kinase (CK), which leaks out of muscles that are deteriorating. This is a nonspecific test because CK levels are elevated in many neuromuscular diseases, but it is often useful anyway.
Genetic testing can be performed if SMA is suspected, because this is the least invasive and most accurate way to identify chromosome 5-related SMA of all types. Both capillary electrophoresis (CE) and next generation sequencing (NGS) methodologies are used in currently available kits. Tests are offered by a number of companies including Centogene, Arup and several US and Canadian based organisations.
A new microarray-based carrier assay includes the mutations for SMA as well as mutations for around 600 other inherited diseases, in a single blood-based assay (fig 3).

Figure 3 CarrierScan 1S assay: a single solution for expanded carrier screening research
It includes an analysis for SMN1 status as part of its SMN Reporter module, with easy visualization and summary of copy number states of SMN1 Automated exports are generated in the form of tab-delimited text files and PNG graphic files that are amenable to downstream applications
SMN Reporter
SMN Reporter includes:
- Novel algorithms to determine SMN1 based carrier status
- Graphical depiction of SMN copy number
- Support for automated single sample visual exports

Get more information on the Applied Biosystems CarrierScan assay.
Leave a Reply