がんドライバー遺伝子および炎症
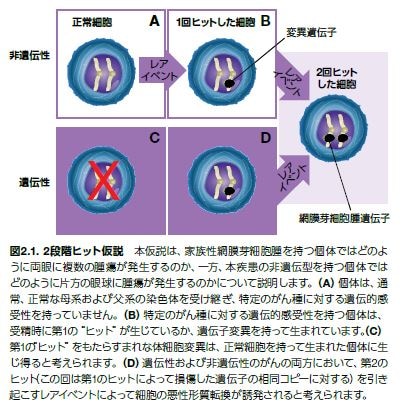
1940年代から1950年代に研究者は、がんの発症には遺伝的背景が関与している可能性があると仮定し始めました*1 。そのような考えは、現代の分子がん研究を立ち上げたパラダイムシフトを代表するものです。1950年代の間は疫学データおよび数学的モデルに依存していましたが、Nordlingらの研究者は、がんは連続的な変異(すなわちヒット)によって発生すると提唱しました。これにより、高齢化に伴うさまざまなヒトがんの発生頻度の増加を説明することが可能です*2 。Knudsonは、がんの発症に関与するヒットの最小数について理解するために、通常幼児期に発症する網膜の腫瘍である網膜芽細胞腫(RB)の根底にある遺伝的メカニズムについて調査する研究を開始しました(図2.1)*3 。Knudsonの統計解析から、疾患には2つのヒットまたは変異が関与する2種類の形態が存在することが予測されました。遺伝性網膜芽細胞腫のケースでは、最初のヒットは受精時に存在する遺伝子変異に関係していたのに対し、それに続く第2のヒットは同じ遺伝子の他方のコピーに生じる体細胞変異でした。家族性網膜芽細胞腫を受け継いだ子供が第2のヒットを生じる可能性は、変異を持たない子供の100,000倍以上です。逆に言えば、このモデルから、網膜芽細胞腫を生じる遺伝子を持たない個体において疾患が生じるには、2つの体細胞変異(類似した変異率で発生する)が必要であることが予測されました*4。
1971年に、間接的にがん抑制遺伝子の概念につながる研究が公開され、発がんの2段階ヒット仮説(Knudson仮説)が提唱されました。これは、網膜芽細胞腫を含む数種の主要ながんにおいて見られる変異であるRB1 遺伝子が発見される15年前で、初めてヒト染色体の配列決定が行われた約28年前のことでした*5,6。
家族性がん
RB1遺伝子変異の他に、さまざまなヒトがん抑制遺伝子が家族性がん症候群および非遺伝型のがんと関連します。新しい候補遺伝子が同定されると順次、調査および検証されていくものと考えられます*7。
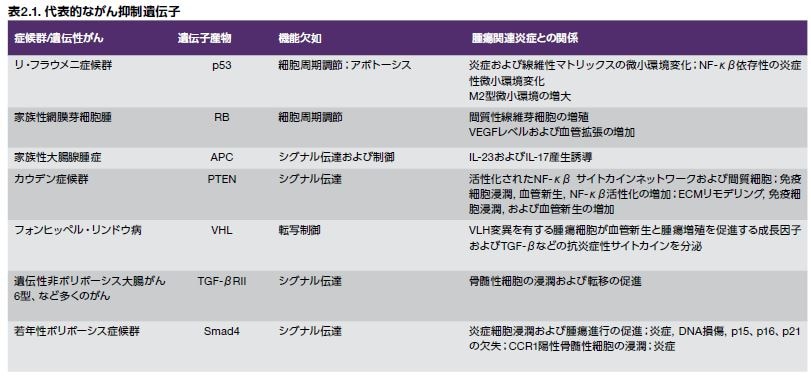
RB1遺伝子の変異に関連するがんの高い発生率は、このがん抑制遺伝子が重要であることを浮き彫りにしています。RB1の機能喪失は、小児における網膜芽細胞腫に関係します。RB1遺伝子によってコードされるタンパク質であるpRbは、細胞周期の制御因子であり、RB経路の調節不全はヒトがんの大部分の形態において観察されます。*8 家族性がんおよび散発性がんに関連するがん抑制因子の例を表2.1に示します*9。
![Rb[pSpT249/252] ABfinity™ Recombinant Rabbit Monoclonal Antibody](https://www.thermofisher.com/blog/wp-content/uploads/sites/13/2019/07/1-34.jpg)
がん抑制機能喪失
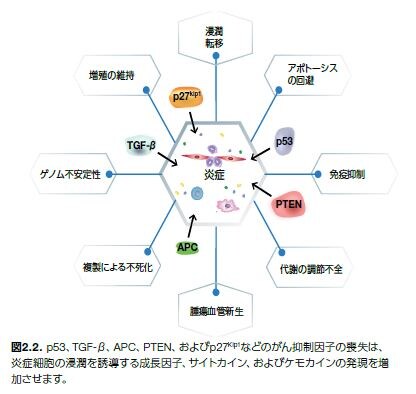
がん抑制機能の喪失は、腫瘍形成を誘導する炎症過程に寄与することも知られています。がん抑制遺伝子は、ストレスおよびDNA損傷に対する細胞応答の制御や、細胞周期、老化、およびアポトーシスに関連するプロセスの調節に不可欠です。がん抑制因子の関与が、がん関連炎症反応の調節に役割を果たしていることは複数の実験的証拠で明らかにされています*10。
- p53によるがん抑制機能喪失は、潰瘍性大腸炎罹患患者における結腸直腸がんのリスク増加に関与
- APCによるがん抑制機能喪失は、腸において腫瘍形成を誘導するIL-17産生増進に関与
- TGF-βシグナル伝達の喪失は、炎症細胞の腫瘍微小環境への流入に関与
腫瘍関連炎症に関わる既知のがん抑制因子の例を図2.2に示します。また、数種の腫瘍抑制因子遺伝子が炎症の誘導に寄与する役割に関する一般情報を表2.1に示します*11。
がん抑制因子TP53 をコードする遺伝子における体細胞変異は、ヒトゲノムにおけるいずれの他の既知のドライバー遺伝子に影響を及ぼす変異よりも高頻度で発生します。p53シグナル伝達の調節不全は、無制御な細胞増殖を引き起こします。また、TP53における生殖細胞系列の異常は、リ・フラウメニ症候群―さまざまながんの早期発症の素因がある状態―を生じます。p53に関する情報は、1979年に初めて発表され、その数十年後に遺伝子の発がん特性について確認されました*12。
野生型p53
正常なホメオスタシスを維持するために、DNA損傷、低酸素、およびがん遺伝子活性化によって野生型p53が活性化されます。野生型p53―配列特異的転写因子―は、細胞周期の進行を阻害したり、老化を促進したり、あるいはストレッサーに対する反応においてアポトーシス細胞死を誘導します。がん抑制遺伝子の中で、TP53は最も広範に研究されており、多様なヒト腫瘍においてp53の不活性化が実証されています。TP53における有害突然変異は、異常な細胞増殖、ゲノム不安定性、およびがん進行につながります*11,13。
p53の他の機能
前述のがん抑制因子としての機能の他にも、p53関連機能について実験的証拠が得られています。正常状態下で、p53は、炎症反応の調節など、複数の細胞プロセスに関連する遺伝子を転写的に制御しています(図2.3)*13-17。
- リボソーム生合成
- 老化の制御
- p53標的遺伝子の転写制御による自然免疫系の発現増加
- IFN調節因子(IRF)IRF-9およびIRF-5(抗ウイルス反応)
- Toll様受容体3(TLR3)によるウイルス感染の認識およびIFN経路の誘導を介した抗ウイルス応答の活性
- 単球走化性タンパク質-1(MCP-1)
単球-マクロファージ系統の細胞の動員および活性化
- 自然免疫系および獲得免疫系のさまざまな細胞の調節に関連する遺伝子の転写制御
潰瘍性大腸炎(UC)関連結腸直腸がんの発症におけるp53の不活性化の関連性については明確にされています。UC患者の非がん結腸組織におけるTP53変異の頻度の増加は、DNA損傷、組織損傷を引き起こす活性酸素種(ROS)および一酸化窒素(NO)の活性を上昇させ、腫瘍形成を促進します。TP53の変異は、散発性結腸直腸がんの進行においても観察されます*18 。TP53変異と、炎症反応の増加、および腫瘍形成との関係性は、頭頸部、乳房、肝臓、および他の組織の腫瘍を含む他のがん形態でも説明されています*11。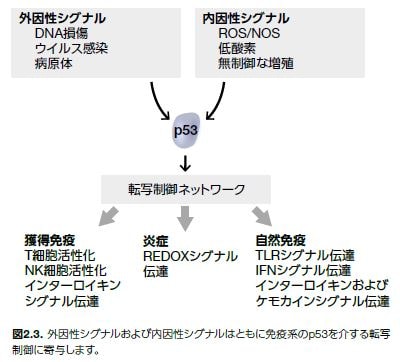
当記事は、腫瘍関連炎症を評価するための抗体ベースのツールガイドブックからの抜粋です。
ガイドブックは下記から無料でダウンロード頂けます。
Adenomatous polyposis coli (APC)
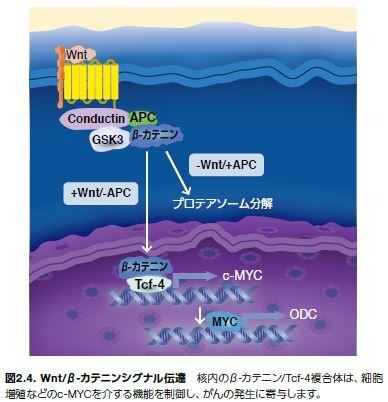
Wnt/β-カテニン経路の制御機構は非常に複雑です。Wntおよびβ-カテニンはともにDrosophila(ショウジョウバエ)属で最初に発見された、一連の核となる発生関連シグナル伝達タンパク質の一部です*19 。Wnt経路シグナル伝達の重要な制御因子として、APCはconductinを含む構造成分の細胞間結合に関与し、α-およびβ-カテニンの特定の内部領域への結合においてE-カドヘリンと競合します。APCは、活性型β-カテニン/転写因子4(TCF4)複合体の形成を制御します。APCは、明確になっているがん抑制因子で、APCタンパク質の喪失または減少につながる生殖細胞系列変異は大腸および直腸のがんに関与する家族性大腸腺腫症(FAP)に関連します。APCにおける体細胞変異は、肺、乳房、結腸、および他の器官の悪性腫瘍にも関連します*20,21。
APCの欠失および腫瘍関連炎症
APCは、腸上皮細胞の増殖、および接着の制御因子です。APCの遊走および生殖細胞系列アレル欠失は、ヒト家族性大腸腺腫症症候群、および散発性結腸直腸がんのホールマークです。結腸直腸腫瘍は、免疫/炎症性浸潤および炎症性遺伝子特性を示します。APCは結腸がんのホールマークであることから、これらの臨床研究においてAPCと炎症に関連がある可能性が示唆されています。複数の実験的証拠において、結腸がんの進行におけるAPC欠損と腫瘍関連炎症の関与が示されています。これらの知見の代表例をいくつか示します*22,23。
- 結腸直腸がんのマウスモデルにおいて、APC制御の喪失はその後の炎症誘発性可溶性因子のレベル増加と相関
変異型APCアレルを持つマウス腸上皮細胞における野生型APCアレルの不活性化は、腫瘍形成を促進します。 - IL-23(微生物産物によるAPC欠損腺腫の浸潤に反応して、腫瘍浸潤骨髄性細胞において発現)
IL-17の発現を促進します。 - IL-17Aは、特定のT細胞および自然免疫系の細胞によって産生します。
- IL-17の上方制御は、結腸直腸がんの進行および予後不良と相関します。
- シクロオキシゲナーゼ-2(COX-2)は、腸上皮細胞の悪性形質転換を促進するプロスタグランジンE2の産生に不可欠です
アスピリンなどの非ステロイド性抗炎症薬(NSAID)は、強力な予防効果を示し、近年、結腸直腸がんの補助療法として提案されています*11。
ホスファターゼおよびテンシンホモログ(PTEN)
腫瘍抑制因子ホスファターゼおよびテンシンホモログ(PTEN)は、脂質およびタンパク質のデュアルホスファターゼ活性を持ち、ホスファチジルイノシトール3-キナーゼ(PI3K)を介するシグナル伝達の活性を抑制することによって、Aktを阻害します。PTENは、脂質ホスファターゼとして機能し、PIP3のイノシトール環の3位の水酸基を脱リン酸化し、PIP2を生成します。PTENは、PI3K/Akt/mTORシグナル伝達カスケードの負の制御因子として、増殖、生存、およびエネルギー代謝などの基本的な細胞機能の制御において極めて重要な役割を果たしています(図2.5)*24,25。 PTENは、PIP3と相互作用する他に、PI3KおよびFAKなどのチロシンリン酸基を持つ特定のタンパク質も認識し、シグナル伝達タンパク質Shcなどのセリン残基およびスレオニン残基を持つ特定のタンパク質に対するホスファターゼとしても作用します*26。
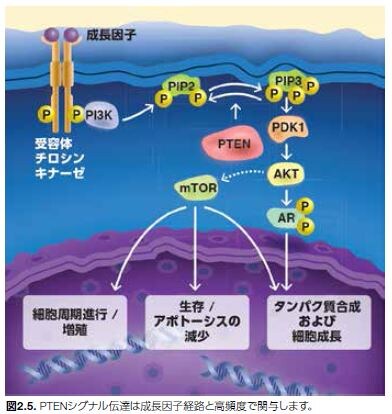
 数多くの臨床所見および実験所見において、腫瘍関連炎症の阻害においてPTENの果たす役割が支持されています。これらの知見を以下に示します*11,27。
数多くの臨床所見および実験所見において、腫瘍関連炎症の阻害においてPTENの果たす役割が支持されています。これらの知見を以下に示します*11,27。
- ヒト頭頸部扁平上皮がんにおいて、PTEN制御の喪失は、単球浸潤および腫瘍血管新生の誘導およびNF-κBの上方制御と相関します。
- がん遺伝子KRASによって誘導された膵管腺がんのマウスモデルにおいて、PTEN機能の喪失は、NF-κB活性の上方制御、間質活性化、および炎症細胞の流入と相関します。
- PTEN機能の喪失は、炎症促進性反応を誘導し、乳がんおよび前立腺がんの進行を促進することも報告されています。
トランスフォーミング増殖因子β(TGF-βRII)
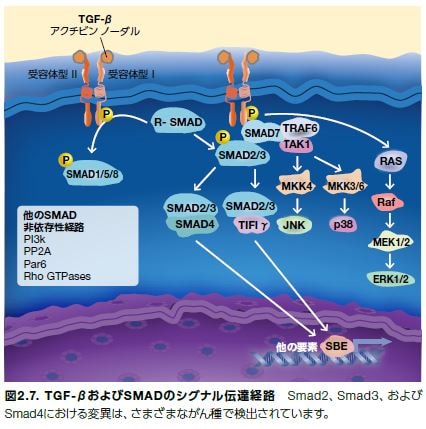
トランスフォーミング増殖因子β(TGF-β)は、構造的にホモ二量体を形成するサイトカインのファミリーに属します。このファミリーにはアクチビンおよび骨形態形成タンパク質も含まれます。哺乳類アイソフォームには、TGF-β1、TGF-β2、およびTGF-β3があり、関連するセリン/スレオニン受容体とともに、これらに限定されませんが、細胞増殖、血管新生、免疫反応、およびアポトーシスなどの複数の生物学的プロセスを制御しています。TGF-βシグナル伝達の調節不全は、創傷治癒の異常、組織線維化、心血管疾患、自己免疫疾患、およびがんを含む他の障害に関係します。Smadは、進化的に保存された細胞内タンパク質で、セリン/スレオニンキナーゼ受容体によって活性化され、核移行が生じ、それに続きTGF-βシグナル伝達の転写制御因子として機能します*28,29。

腫瘍抑制におけるTGF-βシグナル伝達
複数の研究において、腫瘍抑制におけるTGF-β経路メディエーターの関与が示されています*30–32 。さまざまな腫瘍において、TGF-β受容体およびSmadタンパク質をコードする遺伝子における孤発性変異が同定されています*33,34 。また、TGF-βRIIをコードする遺伝子における機能喪失型の生殖細胞系列変異は、遺伝性非ポリポーシス結腸直腸がん(HNPCC)に関係します。TGF-βRIIにおける不活性化変異は、結腸がんの20~25%、ならびに胃、膵臓、乳房、肝臓、および他の器官のがんにおいても高頻度で発生していることが知られています。TGF-βR1をコードする遺伝子の不活性化変異についても報告されており、卵巣、乳房、膵臓、およびTリンパ球のがんを誘導することが知られています。複数の研究において、腫瘍はTGF-β受容体の発現を高頻度で下方制御し、TGF-βによる増殖阻害から回避していることが示されています*30,35,36 。TβRIIの腫瘍抑制因子としての根拠は、以下の代表例によって支持されています:
- 野生型TβRIIの外因性発現は、内因性TGF-βRII機能活性が欠如した結腸、乳房、または甲状腺のがん細胞において腫瘍の増殖を抑制しました。
- トランスジェニックマウスの皮膚におけるTGF-β1またはTβRIIの過剰発現は、がん抑制因子としての根拠も提供しました。
- マウス乳腺および肺におけるドミナントネガティブ変異型TβRIIのトランスジェニック過剰発現は、化学発がん物質に反応した腫瘍形成を増加させました。
TGF-β経路には、腫瘍抑制活性の他に、腫瘍促進活性もありますが、この二面性については解明されていません。例えば、いくつかの体細胞変異を持たない上皮がんは、TGF-βの増殖抑制作用を克服します*37。
TGF-βシグナル伝達および炎症
さまざまな研究において、TGF-βシグナル伝達の消失が腫瘍浸潤性炎症細胞の存在を増加させることがで示されています。また、複数の臓器の上皮細胞におけるTGF-βRII変異は、炎症誘発性状態を促進することによる腫瘍進行と相関しています。さらに、がん関連線維芽細胞におけるTGF-β機能の喪失は、炎症促進性反応に必要とされる遺伝子の発現を増進し、腫瘍進行に寄与しました*11。
Smad4
ヒトSmad4は、8種類のヒトSmadアイソフォームの一つで、TGF-βRの下流で機能します。Smadタンパク質は、共通の構造的特徴を持ちますが、3種類の異なる機能的クラスに分類されます:*38,39
- 受容体制御型Smadタンパク質(R-Smad)は、TGF受容体によって直接リン酸化され、活性化されます。
̶ Smad1、Smad5、およびSmad8(BMP シグナル伝達を媒介)
̶ Smad2、Smad3(TGF-β/アクチビン シグナル伝達を媒介) - 共有型Smadは、R-Smadと複合体を形成し、核移行を経て、標的遺伝子の転写制御因子として機能します。
̶ Smad4 - 抑制(拮抗)型Smadまたは抗-Smadは、シグナル伝達の負の制御因子として機能します。
̶ Smad6(グローバルな TGF-β経路の阻害剤)
̶ Smad7(BMP-特異的経路の阻害剤)
当記事は、腫瘍関連炎症を評価するための抗体ベースのツールガイドブックからの抜粋です。
ガイドブックは下記から無料でダウンロード頂けます。
腫瘍抑制因子としてのSmad4
Smad4の腫瘍抑制因子機能についてはすでに立証されています。膵がんおよび結腸直腸がんは、高頻度でSmad4変異を持ちます。Smad4機能の喪失は、非がん性ポリープの存在および消化(GI)管における悪性腫瘍発現リスクの増加を特徴とする遺伝性疾患である若年性ポリポーシス症候群に関連します。また、Smad4調節不全は、乳がんの腫瘍形成および転移に関与していることも示されています。結腸上皮細胞におけるSmad4混乱が腫瘍進行に寄与する炎症性骨髄細胞の動員を増加させることについてはさまざまな報告があります。Tリンパ球におけるSmad4欠失は、GIがんの発症に寄与する炎症誘発性サイトカインIL-5、IL-6、およびIL-13の分泌を誘導します*40,41。
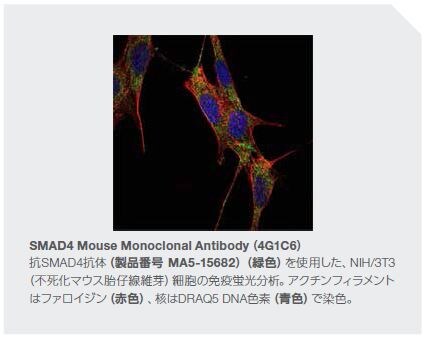
Smad4と同様、Smad2およびSmad3は、腫瘍抑制因子として同定されています。しかしながら、低レベルのSmad3は腫瘍の抑制よりも進行を促進するという報告があります*42,43。
がんのドライバー変異およびパッセンジャー変異
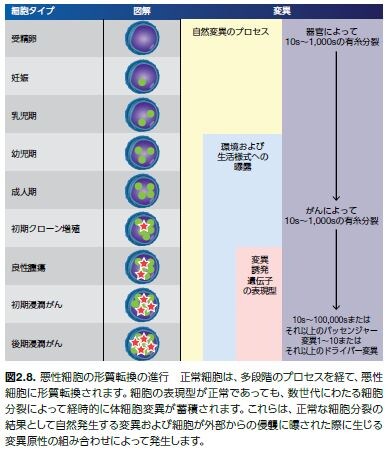
がん細胞で検出される点変異および転座において、遺伝子変異の大部分は腫瘍細胞の適応性(すなわち選択的増殖優位性)に影響しません(図2.8)*44,45。 周囲の正常細胞よりも腫瘍細胞に対し増殖優位性を与えることがない変異はパッセンジャー変異と定義されます。一方、周囲の正常細胞よりも腫瘍細胞に対し増殖優位性を与える遺伝子はドライバー変異と考えられます。ドライバー遺伝子の中には、ドライバー変異が含まれます。しかしながら、ドライバー遺伝子にはパッセンジャー変異も含まれ得ることに留意することは重要です*46,47。
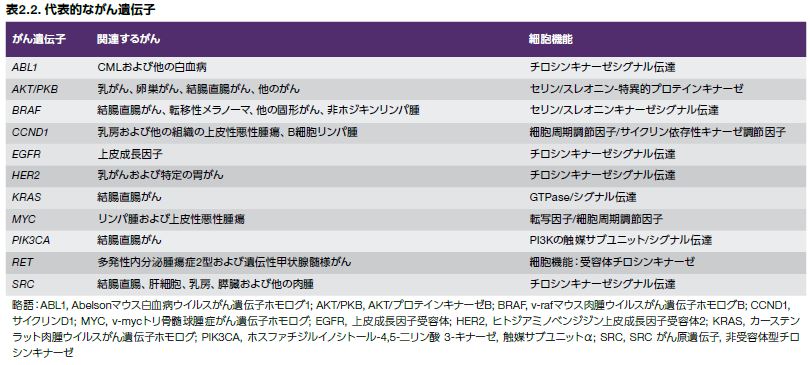
 腫瘍抑制変異と同様に、発がん性変異はドライバー変異として分類されます。がん遺伝子は、正常な細胞プロセスを調節するがん原遺伝子に由来しますが、がん原遺伝子における機能獲得型変異は、腫瘍への形質転換および増殖を誘発する可能性を持つ異常な遺伝子産物を産生します(図2.9)。(代表的ながん遺伝子については、表2.2を参照してください)。
腫瘍抑制変異と同様に、発がん性変異はドライバー変異として分類されます。がん遺伝子は、正常な細胞プロセスを調節するがん原遺伝子に由来しますが、がん原遺伝子における機能獲得型変異は、腫瘍への形質転換および増殖を誘発する可能性を持つ異常な遺伝子産物を産生します(図2.9)。(代表的ながん遺伝子については、表2.2を参照してください)。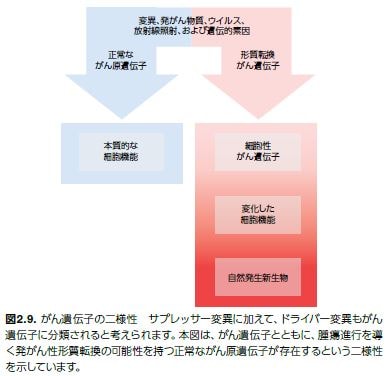
遺伝子変異の蓄積は、最終的に細胞周期調節に必要とされるメカニズムを妨害します。正常な状態では、増殖シグナルの産生および伝播ならびに細胞増殖が高度に調節されています。増殖プロセスの厳密な制御は、健全な組織の構造および機能の維持をサポートします。増殖に関連するシグナル伝達経路は、通常、必要になると活性化され、不要になると不活性化されます。悪性細胞のホールマークの一つは、恒常的調節の喪失、および持続的、あるいは制御不能な増殖です*49。
がん遺伝子および腫瘍関連炎症
がん遺伝子変異は、悪性細胞形質転換のイニシエーターとしてのがん遺伝子の役割が十分に立証されている他、腫瘍の炎症の誘導に関与し、炎症誘発性メディエーターおよび血管新生メディエーターの産生を導きます。例えば、RASがん遺伝子のアイソフォームHRASおよびKRASにおける変異は、NF-κBならびにヒトがん細胞における炎症誘発性サイトカインおよびケモカインの産生を増進します。同様に、がん遺伝子BRAFにおける変異は、メラノーマにおける炎症プログラムとの関係性が指摘されています。また、MYC、RET、およびEGFRなどの遺伝子における発がん性変異は、さまざまな固形がんにおける腫瘍関連炎症の誘導をもたらしています*50。
RETがん遺伝子および炎症
RETがん遺伝子シグナル伝達経路については、がん化シグナル伝達と腫瘍形成を促進する炎症反応との関係についての十分に確立された例を提供します。RETがん原遺伝子は、GDNFファミリーリガンドと相互作用する受容体チロシンキナーゼをコードします。RET機能喪失変異は、ヒルシュスプルング病に関連し、発がん性変異は内分泌系および甲状腺に発生するがんを引き起こします。例えば、甲状腺細胞の形質転換は、RETがん遺伝子の染色体再編によって甲状腺乳頭がんに進行する可能性があり、特定の点変異は甲状腺髄様がんに関係します。RETにおける変異は、甲状腺細胞の機能を調節する炎症メディエーターをコードする遺伝子の調節不全も誘導する可能性があります。̶これらのタンパク質の中には、成長因子、ケモカイン受容体、サイトカイン、メタロプロテアーゼ、および他のタンパク質クラスが含まれます(図2.10および表2.3)。がんの発生におけるこれらの因子の特定の役割については調査中です。RETの活性化変異を持つ甲状腺がんの治療には数多くの低分子阻害剤が使用されています。̶その多くが甲状腺がん治療薬として食品医薬品局(FDA)に承認されています*51,52。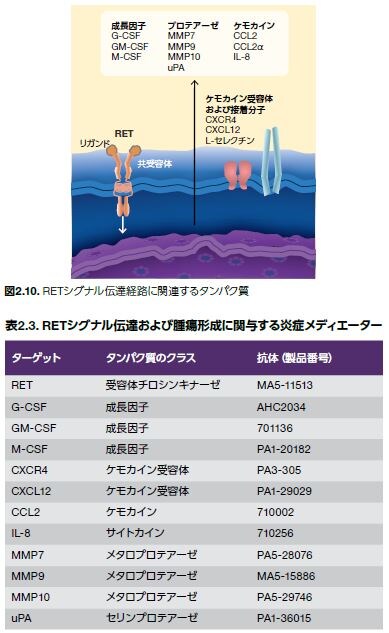
炎症および腫瘍進行
腫瘍抑制機能喪失とがんの進行に寄与する炎症の発生における発がん活性との関連性については、数多くの研究において明らかにされています。感染、化学刺激、および組織損傷などの因子は急性炎症につながります。また、遺伝的不安定性(変異)は、炎症細胞および間質細胞の動員、浸潤、および活性化を特徴とする局所炎症反応にさらに寄与するケミカルメディエーター発現の上昇につながります。炎症が未解決である場合、慢性炎症は腫瘍促進に寄与します。すなわち、始原細胞から良性病変が発生し腫瘍に進行するプロセス、良性腫瘍が悪性腫瘍に進行するプロセスをサポートします(図2.11)。腫瘍発生における感染、炎症、および遺伝的不安定性の関係についてサポートするデータは豊富にありますが、がんに関連する炎症の正確なメカニズムについては完全には理解されていません。しかしながら、これらの発がん促進イベントの中核をなすのは、マクロファージ、それらの炎症メディエーター、ならびに他の免疫系の細胞の存在です*53。
腫瘍微小環境は高度に複雑で、がんの促進と抑制の両方の機能を持つ細胞の集合で構成されます。研究者は、膨大なリソースを費やして、腫瘍微小環境に寄与する細胞の複雑なネットワークおよび可溶性因子を制御するプロセスの解明を目指して尽力しています。腫瘍微小環境内の炎症誘発性状態を減少させるためのアプローチを考案および試験し、腫瘍拒絶を増強する可能性を有する殺腫瘍性リンパ球およびNK細胞などの細胞の活動を増強する状態を誘導するための取り組みが進められています*54。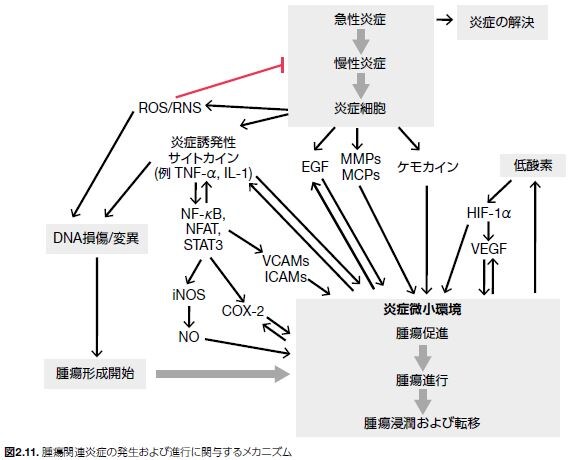
抗体: 研究のための強力なツール
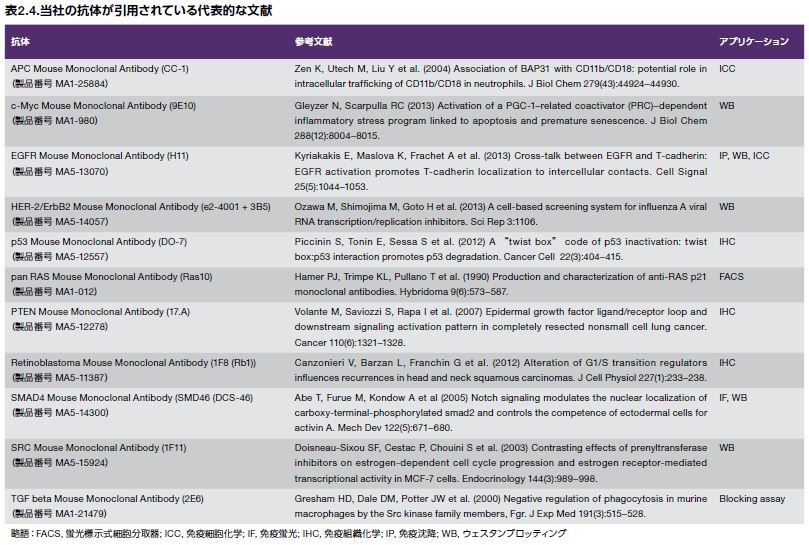
高品質の生物医学研究用抗体を生産する能力は、年を追うごとに がん生物学分野における新しい発見に貢献しています。当社では、数千ものがん研究用の一次抗体および二次抗体を提供しています。お客様の研究のフォーカスが増殖シグナル伝達、転移、アポトーシス、オートファジー、代謝、炎症、腫瘍抑制因子、または他のどのようながん関連研究分野であっても、当社はお客様の研究の成功を確実にする研究用抗体の幅広いセレクションを取りそろえています。当社の抗体アッセイが幅広い抗体アプリケーションにおいて優れた実験結果の達成に寄与していることは、世界中の数千もの文献引用により実証されています。
抗体検索ツールで、目的の研究に適した抗体を見つけるには、こちらをご覧ください。
【無料ダウンロード】腫瘍関連炎症を評価するための抗体ベースのツール
当記事は、腫瘍関連炎症を評価するための抗体ベースのツールガイドブックからの抜粋です。以下の内容を含むPDFは下記から無料でダウンロード頂けます。
- 慢性炎症およびがん
- がんドライバー遺伝子および炎症
- 腫瘍微小環境
- シグナル伝達および腫瘍関連炎症
- 要約および付録
【無料ダウンロード】がん研究を促進させる遺伝子解析技術ハンドブック
また、複数のがん研究領域における興味深い最新の発見を可能にする遺伝子解析技術を紹介するハンドブックも配布しています。併せてごらんください。
参考文献:
1. Frank SA Dynamics of cancer: incidence, inheritance and evolution. Chap. 4Principal of theories. Princeton (NJ): Princeton University Press; 2007. Available: http://www.ncbi.nlm.nih.gov/books/NBK1569/. Accessed March 2015.
2. Marte B (2006) Milestone 9 (1953) Two-hit hypothesis. It takes (at least) two to tango. Nature Milestones Cancer. Available: http://www.nature.com/milestones/milecancer/full/milecancer09.html. Accessed March 2015.
3. Fox Chase Cancer Center. Alfred G. Knudson Jr. summary page.https://www.foxchase.org/blog/2013-04-30-dr-knudson-and-the-two-hit-theory. Accessed March 2015.
4. Knudson AG Jr. (1971) Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA 68(4):820–823.
5. National Human Genome Research Institute. Scientists complete first chapter of book of life with decoding of first human chromosome.1999. Available: http://www.genome.gov/10002104. Accessed March 2015.
6. Chial H Tumor suppressor (TS) genes and the two-hit hypothesis. Nature Education. 1(1):177. Available: http://www.nature.com/scitable/topicpage/Tumor-Suppressor-TS-Genes-and-the-Two-887. Accessed March 2015.
7. Weinberg R The Biology of Cancer, 2nd ed. New York (NY): Garland Science; 2014. Available: https://books.google.com/books?id=MzMmAgAAQBAJ&pg=PA269&lpg=PA269&dq=Human+tumor+suppressor+genes+that+have+been+cloned&source=bl&ots=A0JkRa800f&sig=kKyxZPicikTAlDZL6ImeHvhiicI&hl=en&sa=X&ei=rZ-a1VPDMD4L9yQSb9oHACQ&ved=0CDIQ6AEwAjgK#v=onepage&q=Human%20tumor%20suppressor%20genes%20that%20have%20been%20cloned&f=false
8. Goodrich DW (2006) The retinoblastoma tumor-suppressor gene, the exception that proves the rule. Oncogene 25(38):5233–5243.
9. The Medical Biochemistry Page (2015). Tumor suppressor genes and activities. http://themedicalbiochemistrypage.org/tumor-suppressors.php
10. Través PG, Luque A, Hortelano S (2012) Macrophages, inflammation, and tumor suppressors: ARF, a new player in the game. Mediators Inflamm 2012:568783.
11. Yang L, Karin M (2014) Roles of tumor suppressors in regulating tumor-associated inflammation. Cell Death Differ 21(11):1677–1686.
12. Olivier M, Hollstein M, Hainaut P (2010) TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol 2(1):a001008.
13. Lowe J, Shatz M, Resnick MA et al. (2013) Modulation of immune responses by the tumor suppressor p53. Biodiscovery 8:2.
14. Fuster JJ, Sanz-González SM, Moll UM et al. (2007) Classic and novel roles of p53: prospects for anticancer therapy. Trends Mol Med 13(5):192–199.
15. Munoz-Fontela C, Garcia MA, Garcia-Cao I et al. (2005) Resistance to viral infection of super p53 mice. Oncogene 24(18):3059–3062.
16. Taura M, Eguma A, Suico MA et al. (2008) p53 regulates Toll-like receptor 3 expression and function in human epithelial cell lines. Mol Cell Biol 28(21):6557–6567.
17. Huang S, Singh RK, Xie K et al. (1994) Expression of the JE/MCP-1 gene suppresses metastatic potential in murine colon carcinoma cells. Cancer Immunol Immunother 39(4):231–238.
18. Hussain SP, Amstad P, Raja K et al. (2000) Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: a cancer-prone chronic inflammatory disease. Cancer Res 60(13):3333–3337.
19. Clevers H, Nusse R (2012) Wnt/β-catenin signaling and disease. Cell 149(6):1192–1205.
20. Genetics Home Reference. Familial adenomatous polyposis. Available: http://ghr.nlm.nih.gov/condition/familial-adenomatous-polyposis. Accessed March 2015.
21. Virmani AK, Rathi A, Sathyanarayana UG et al. (2001) Aberrant methylation of the adenomatous polyposis coli (APC) gene promoter 1A in breast and lung carcinomas. Clin Cancer Res 7(7):1998–2004.
22. Polakis P (2012) Wnt signaling in cancer. Cold Spring Harb Perspect Biol 4(5).
23. Grivennikov SI, Wang K, Mucida D et al. (2012) Adenoma-linked barrier defects and microbial products drive IL-23/IL-17–mediated tumour growth. Nature 491(7423):254–258.
24. Jabbour E, Ottmann OG, Deininger M, et al. (2014) Targeting the phosphoinositide 3-kinase pathway in hematologic malignancies. Haematologica 99(1):7–18.
25. Song MS, Salmena L, Pandolfi PP (2012) The functions and regulation of the PTEN tumor suppressor. Nat Rev Mol Cell Biol 13(5):283–296.
26. Gu J, Tamura M, Pankov R et al. (1999) Shc and FAK differentially regulate cell motility and directionality modulated by PTEN. J Cell Biol 146(2):389–403.
27. Garcia AJ, Ruscetti M, Arenzana TL et al. (2014) Pten null prostate epithelium promotes localized myeloid-derived suppressor cell expansion and immune suppression during tumor initiation and progression. Mol Cell Biol (11):2017– 2028.
28. Akhurst RJ, Hata A (2012) Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discov 11(10):790–811.
29. Reinder J, Datta P (2011) TGF-beta-dependent and -independent roles of STRAP in cancer. Frontiers Biosci 16:105–115.
30. Derynck R, Akjurst J, Balmain A (2001)TGF-beta signaling in tumor suppression and cancer progression. Nat Genet 29(2):117–129.
31. Pardali K, Moustakas A (2007) Actions of TGF-beta as tumor suppressor and pro-metastatic factor in human cancer. Biochim Biophys Acta 1775(1):21–62.
32. Antony ML, Nair R, Sebastian P et al. (2010) Changes in expression, and/or mutations in TGF-beta receptors (TGF-beta RI and TGF-beta RII) and Smad 4 in human ovarian tumors. J Cancer Res Clin Oncol 136(3):351–361.
33. de Caestecker MP, Piek E, Roberts AB (2000) Role of transforming growth factor-beta signaling in cancer. J Natl Cancer Inst 92(17):1388–1402.
34. Xu Y, Pasche B (2007) TGF-beta signaling alterations and susceptibility to colorectal cancer. Hum Mol Genet 16 Spec No 1:R14–20.
35. Shin KH, Park YJ, Park JG (2000) Mutational analysis of the transforming growth factor beta receptor type II gene in hereditary nonpolyposis colorectal cancer and early-onset colorectal cancer patients. Clin Cancer Res 6(2):536– 540.
36. Bierie B, Moses HL (2010) Transforming growth factor beta (TGF-beta) and inflammation in cancer. Cytokine Growth Factor Rev 21(1):49–59.
37. Bachman KE, Park BH (2005) Dual nature of TGF-beta signaling: tumor suppressor vs. tumor promoter. Curr Opin Oncol 17(1):49–54.
38. Hata A, Lagna G, Massagué J (1998) Smad6 inhibits BMP/Smad1 signaling by specifically competing with the Smad4 tumor suppressor. Genes Dev 12(2):186–197.
39. Yan X, Liu Z, Chen Y (2009) Regulation of TGF-beta signaling by Smad7. Acta Biochim Biophys Sin 41(4):263–272.
40. Deckers M, van Dinther M, Buijs J et al. (2006) The tumor suppressor Smad4 is required for transforming growth factor beta–induced epithelial to mesenchymal transition and bone metastasis of breast cancer cells. Cancer Res 66(4):2202–2209.
41. Genetics Home Reference. Juvenile polyposis syndrome. Genetics Home Reference. Available: http://ghr.nlm.nih.gov/condition/juvenile-polyposissyndrome. Accessed March 2015.
42. Xu J, Attisano L (2000) Mutations in the tumor suppressors Smad2 and Smad4 inactivate transforming growth factor beta signaling by targeting Smads to ubiquitin-proteosome pathway. Proc Natl Acad Sci USA 97(9):4820–4825.
43. Daly AC, Vizán P, Hill CS (2010) Smad3 protein levels are modulated by Ras activity during the cell cycle to dictate transforming growth factor-beta responses. J Biol Chem. 285(9);6489–6497.
44. Heemskerk B, Kvistborg P, Schumacher TN (2013) The cancer antigenome. EMBO J 32(2):194–203.
45. Stratton MR, Campbell PJ, Futreal PA (2009) The cancer genome. Nature 458(7239):719–724.
46. Vogelstein B, Papadopoulos N, Velculescu VE et al. (2013) Cancer genome landscapes. Science 339(6127):1546–1558.
47. Bozic I, Antal T, Ohtsuki H et al. (2010) Accumulation of driver and passenger mutations during tumor progression. Proc Natl Acad Sci USA 107(43):18545– 18550.
48. Greenman C, Stephens P, Smith R et al. (2007) Patterns of somatic mutation in human cancer genomes. Nature 446(7132):153–158.
49. Chen HZ, Tsai SY, Leone G (2009) Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer 9(11):785–797.
50. Borrello MG, Degl’Innocenti D, Pierotti MA (2008) Infammation and cancer: the oncogene-driven connection. Cancer Lett 267(2):262–270.
51. Mulligan LM (2014) RET revisited: expanding the oncogenic portfolio. Nat Rev Cancer 14(3):173–186.
52. American Cancer Society. Thyroid Cancer. http://www.cancer.org/cancer/thyroidcancer/detailedguide/thyroid-cancer-treating-targeted-therapy
53. Lu H, Ouyang W, Huang C (2006) Inflammation, a key event in cancer development. Mol Cancer Res 4(4):221–233.
54. Shiao SL, Ganesan P, Rugo HS et al. (2011) Immune microenvironments in
研究用にのみ使用できます。診断目的およびその手続き上での使用はできません。
記事へのご意見・ご感想お待ちしています

予測ゲノミクスでがんを理解する
この記事は、科学者向けにがん研究の新しい...
Read More
がん研究を促進するがんオルガノイド(Tumoroid)の培養・解析製品まとめ
がんオルガノイド(Cancer organoid、Tumoroid)は�...
Read More
神経科学におけるフラグメント解析:複数の疑問を解決する柔軟なツール
脳は驚くべき器官です。さらに驚くべきこと...
Read More
リキッドバイオプシーを活用した肺がんと悪性脳腫瘍に対する研究事例のご紹介
リキッドバイオプシーは血液や尿などの体液...
Read More