ホスファチジルイノシトール3-キナーゼ/プロテインキナーゼB/哺乳類ラパマイシン標的タンパク質(PI3K/Akt/mTOR)経路は、広範な正常細胞機能に必要とされる主要なシグナル伝達ノードです。この経路の変化が腫瘍形成の一因であることはよく知られています。なぜなら、この経路は、大部分のがんのホールマーク(代謝、成長および増殖、ならびに転移)の制御に極めて重要で、腫瘍の血管新生やがん関連炎症などの腫瘍促進プロセスにも関与しているからです。PI3K/Akt /mTOR経路は、この経路のいくつかのメディエーターにおいてさまざまな発がん性変異および腫瘍抑制機能喪失につながる変異が同定されたことから、徹底的に探究されてきました。実際、がんゲノム研究では、ヒトがんではこの経路において最も頻繁に変化が生じていることが示されています。
PI3K/Akt /mTORシグナル伝達を制御する細胞機構および分子機構に関する豊富な情報は、経路の阻害剤の開発をサポートし、分子標的抗がん剤の利用可能性の増加に寄与してきました。
▼もくじ [非表示]
PI3K/Akt/mTORシグナル伝達ノード
プロトタイプの生存経路
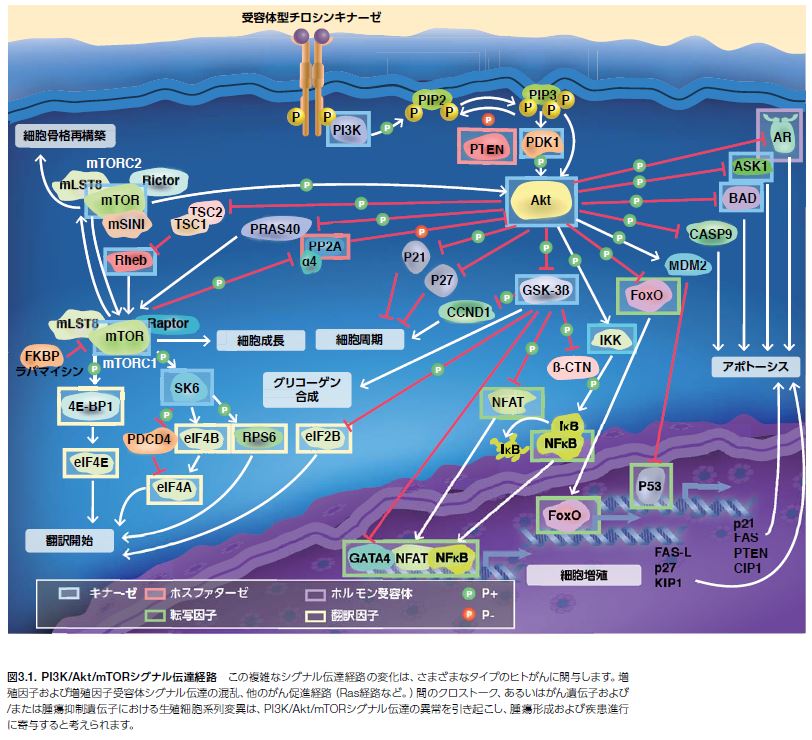
正常なホメオスタシス状態下で細胞機能のさまざまな側面をサポートし、細胞生存に寄与する細胞シグナル伝達経路の変化は、腫瘍形成の進行の一因とされます *1,2。 腫瘍細胞生物学において、ホスファチジルイノシトール3-キナーゼ/プロテインキナーゼB/哺乳類ラパマイシン標的タンパク質(PI3K/Akt/mTOR)カスケードが中心的役割を果たしているという仮定のもと、この基本的なシグナル伝達経路について明らかにするために注力されてきました(図3.1)。多数の研究において、PI3K/Akt/mTOR関連遺伝子に生じた体細胞変異によってさまざまなタイプのがん経路の恒常的活性化が誘導され、腫瘍細胞増殖、成長、分化、代謝、アポトーシス、および腫瘍細胞生存をサポートする他の機能の調節不全が引き起こされている可能性が示されています*3。
PI3K/Akt/mTOR経路に影響する生殖細胞系列の変異は、特定の遺伝性がんに関与します。例えば、腫瘍抑制因子ホスファターゼテンシンホモログ(PTEN)の喪失は、カウデン症候群(多発性過誤腫症候群)を発症させます。この常染色体優性疾患は、皮膚および粘膜の良性病変(過誤腫)の存在を特徴とし、乳房、甲状腺、および他の器官のがんの発生を増加させる可能性があります*4。
ホスファチジルイノシトール3-キナーゼ(PI3K)
PI3Kの機能およびシグナル伝達
ホスファチジルイノシトール3-キナーゼ(PI3K)は、細胞膜の構成要素でホスホイノシチドの代謝前駆体であるホスファチジルイノシトールの3’-水酸基をリン酸化するシグナル伝達タンパク質で、広範な細胞活性を調節することによって細胞生存および細胞死を制御する生物活性脂質分子です(図3.2)*5,6 。PI3Kは、細胞機能の制御において幅広い役割を持つ他、リンパ球の活性化およびシグナル伝達に関与しています*7,8 。
PI3Kは、サイトカイン、インテグリン、B細胞受容体(BCR)活性化、あるいはGタンパク質共役受容体(GPCR)リガンドなどの複数の上流シグナルを介して活性化されます。PI3Kがリガンドに結合すると、受容体チロシンキナーゼ(RTK)-および(GPCR)-誘導性のPI3K活性化が細胞膜で生じます。活性化されたPI3Kは、ホスファチジルイノシトール-4,5- 二リン酸(PtdIns[4,5]P2、またはPIP2)をリン酸化し、ホスファチジルイノシトール- 3,4,5-三リン酸(PtdIns[3,4,5]P3、またはPIP3)を産生させます。続いて、PIP3の蓄積、ならびにAktおよび3-ホスホイノシチド依存性プロテインキナーゼ1(PDK1)などのプレクストリン相同(PH)ドメインを持つタンパク質の動員がシグナル伝達カスケードを誘発し、細胞成長および生存ならびにカルシウム動員、細胞運動性、小胞輸送および細胞増殖、アポトーシス、および他の機能を含む多くのその他の重要な細胞機能に影響を及ぼします(図3.3)*9。
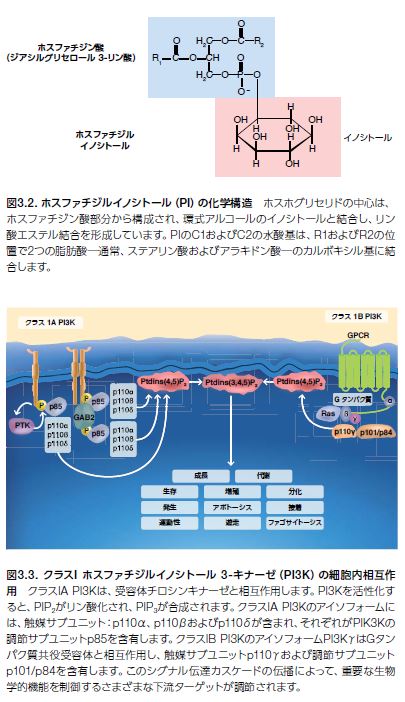 脂質キナーゼのPI3Kには、3種類のクラスが特定されています。図3.3に示したクラスI PI3Kシグナル伝達がヒトがんに果たす役割はよく認識されています。クラスII PI3Kのシグナル伝達(数種のアイソフォームが存在)についてはあまり解明されておらず、ヒト腫瘍進行との関連性は全く知られていません。クラスIII PI3Kで唯一知られているのは、オートファジー―リソソーム内で細胞質成分を分解するシステム―の制御で、腫瘍形成における中心的役割を担っています。PI3Kに影響を及ぼすがんのドライバー変異には、触媒サブユニットの活性化変異やPI3Kヘテロ二量体の調節ドメインに影響を及ぼす機能喪失型変異が含まれます*3 。
脂質キナーゼのPI3Kには、3種類のクラスが特定されています。図3.3に示したクラスI PI3Kシグナル伝達がヒトがんに果たす役割はよく認識されています。クラスII PI3Kのシグナル伝達(数種のアイソフォームが存在)についてはあまり解明されておらず、ヒト腫瘍進行との関連性は全く知られていません。クラスIII PI3Kで唯一知られているのは、オートファジー―リソソーム内で細胞質成分を分解するシステム―の制御で、腫瘍形成における中心的役割を担っています。PI3Kに影響を及ぼすがんのドライバー変異には、触媒サブユニットの活性化変異やPI3Kヘテロ二量体の調節ドメインに影響を及ぼす機能喪失型変異が含まれます*3 。
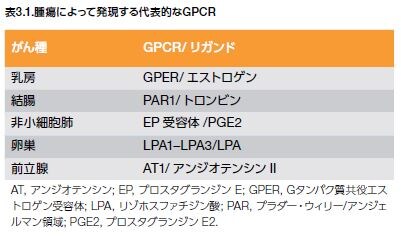
腫瘍細胞進行に関与する一般的な受容体チロシンキナーゼについては第2節で紹介しました。多くのGタンパク質共役受容体は、増殖や腫瘍増殖に寄与する他のプロセスの調節不全と関係していることはよく知られています(表3.1)*10 。

当記事は、がん増殖シグナル伝達経路の探索ガイドブックからの抜粋です。
ガイドブックは下記から無料でダウンロード頂けます。
がんにおけるPI3Kの役割
ヒト腫瘍に関連するPI3K変異
クラスI PI3Kとがんとの関連性については広く報告されており、いくつかのさまざまなタイプのヒト固形がんにおけるPI3K変異の役割が実証されています。さまざまなPIK3CA変異は非ホジキンリンパ腫とも関連しています*11 。
PIK3CA(触媒ドメイン p110α)変異を持つヒト腫瘍タイプ
- 乳房
- 子宮内膜 尿路
- 子宮頸部 皮膚
- 卵巣
- 胃
- 胆道
- 上気道
- 小腸
- 食道
PIK3R1(調節ドメイン)変異を持つヒト腫瘍タイプ
- 子宮内膜 結腸直腸 子宮頸部
- 上気道
- 中枢神経系
- 髄膜
- 乳房
- 食道
- 尿路
- 肺
- 卵巣
- 肝臓
リストの並び順は、PI3K変異の検出頻度が高いがん種から順番に
上から下へ向かって表示しています*3 。
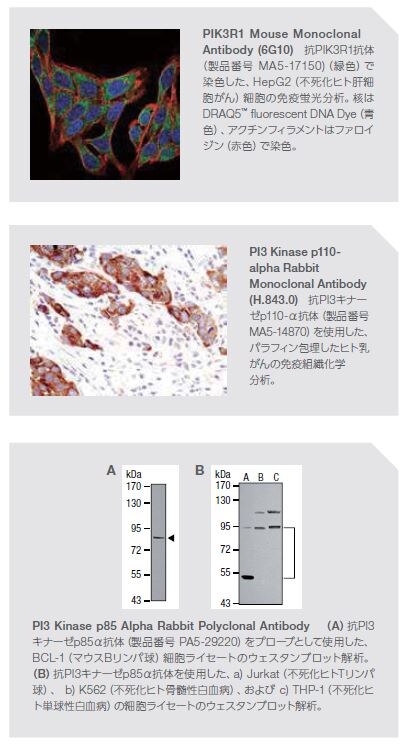 当社は、この主要なシグナル伝達経路のPI3Kおよびメディエーターを検出するための幅広い抗体を提供しています。詳しい情報はこちらをご覧ください。
当社は、この主要なシグナル伝達経路のPI3Kおよびメディエーターを検出するための幅広い抗体を提供しています。詳しい情報はこちらをご覧ください。
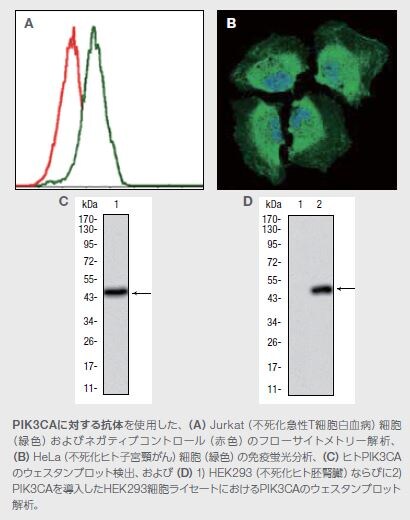
PIK3CA Mouse Monoclonal Antibody (4F3)
製品番号 MA5-17149
アプリケーション:FC、IF、WB
PI3Kは、がん遺伝子として機能する可能性があります。結腸、脳、乳房、胃、肺、および他の器官のがんにおいて、体細胞変異(PI3K遺伝子座における特異的増幅および遺伝子内の欠失)が、PI3K(PIK3CA)の触媒サブユニット110αの活性の上昇を誘導することは多くの研究で確認されています *12 。
ホスファターゼテンシンホモログ(PTEN)
PTENの構造、機能およびシグナル伝達
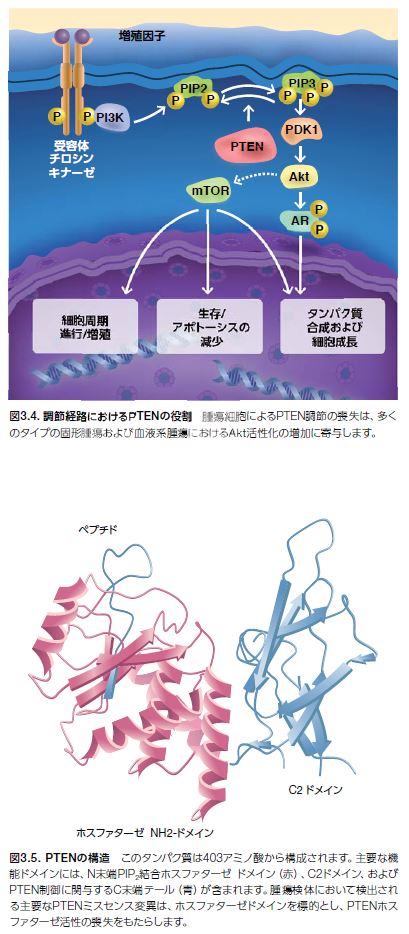
腫瘍抑制因子PTENは、PI3Kを介するシグナル伝達の活性を抑制することによって、Aktを阻害します。PTENは、脂質ホスファターゼとして機能し、PIP3のイノシトール環の3位の水酸基を脱リン酸化し、PIP2を生成します。PTENは、PI3K/Akt/mTORシグナル伝達カスケードの負の制御因子として、増殖、生存、およびエネルギー代謝などの基本的な細胞機能の制御において極めて重要な役割を果たしています(図3.4および3.5)*11,13 。PTENは、PIP3と相互作用する他に、PI3Kおよび接着斑キナーゼ(FAK)などのチロシンリン酸基を持つ特定のタンパク質も認識し、シグナル伝達タンパク質Shcなどのセリン残基およびスレオニン残基を持つ特定のタンパク質に対するホスファターゼとしても作用します*14 。

がんにおけるPTENの役割
PTEN変異を持つヒトがん
- 子宮内膜
- 中枢神経系
- 皮膚
- 結腸直腸
- 前立腺
- 胆道
- 唾液腺
- 眼
- 子宮頸部
- 乳房
- 卵巣
- 胃
PTEN調節の喪失につながる生殖細胞系列変異は、カウデン症候群やバナヤン・ライリー・ルバルカバ症候群などの遺伝性疾患に関係します。これらの状態の影響を受けた個体は、皮膚や身体のさまざまな器官に非がん性病変を発現します。これらの遺伝性疾患は、特定のタイプの悪性腫瘍の発現可能性の増加にも関係します。多数のヒトがんにおいて、ホモ欠失、トランケーション変異、点変異、および他の異常を含む体細胞変異が同定されてきました。PTENは、腫瘍抑制因子p53と同様に、最も一般的ながん関連遺伝子の一つです(図3.5)*3,15 。
がんにおけるPTENの役割
当社は、PTENおよびホスファターゼの直接標的、ならびにPI3K/Akt/mTOR経路が関係する関連タンパク質を検出するための幅広い抗体を提供しています。詳しい情報はこちらをご覧ください。
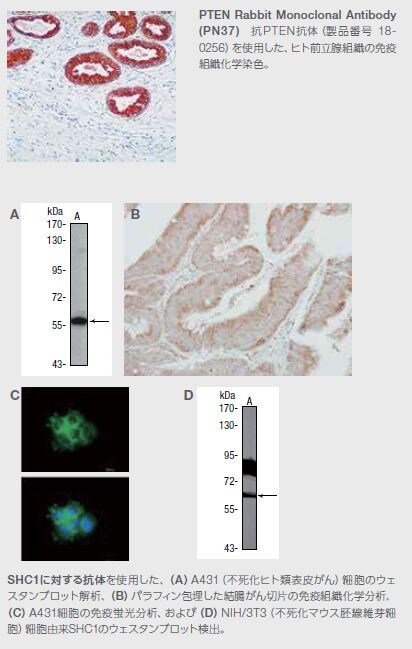
SHC1 Rabbit Polyclonal Antibody
製品番号 PA5-27310
アプリケーション:WB、IHC (P)、 IF
PTENとPI3Kとの相互作用については十分に解明されているのに加えて、アダプター分子SH2ドメインタンパク質C1(Shc1)は、PTENの直接の基質です。3種類の主要なアイソフォームがShc遺伝子によってコードされています。p52Shc およびp46Shcは、増殖因子受容体の下流で機能し、Rasシグナル伝達経路を介するシグナル伝達を促進します*16 。p66Shcアイソフォームは、Shcによる活性酸素種の制御を介する細胞寿命の延長に関係します。いくつかのタイプの固形がんにおいて、Shc1アイソフォームががんの進行に関与している可能性が複数の研究で示されています *17 。詳細な技術プロトコールおよびリソースは付録をご覧ください。
当記事は、がん増殖シグナル伝達経路の探索ガイドブックからの抜粋です。
ガイドブックは下記から無料でダウンロード頂けます。
Akt/プロテインキナーゼB
Aktの構造、機能およびシグナル伝達
セリン/スレオニンキナーゼAkt(プロテインキナーゼBとも呼ばれる)は、広範囲に影響を及ぼすシグナル伝達ネットワークの中心的存在(収束ノード)です。本酵素は、プロテインキナーゼのAGCファミリーのメンバーで、60種類以上のヒトプロテインキナーゼが含まれます。Akt活性化はこれらの細胞シグナル伝達経路のマスタースイッチとして機能し、さまざまな下流ターゲットや相互作用パートナーを介して数多くの細胞内反応を引き起こします。Aktは細胞シグナル伝達における中心的役割を果たし、これらのシグナル伝達の異常はがんおよび糖尿病から神経変性に至る広範囲の疾患に影響を及ぼします。そのため、Aktは、基礎研究および薬剤
開発の両分野において最も積極的に研究されているキナーゼの一つです(図3.6)*19 。
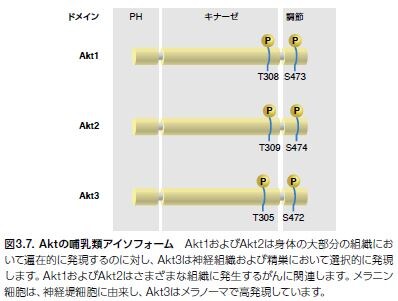
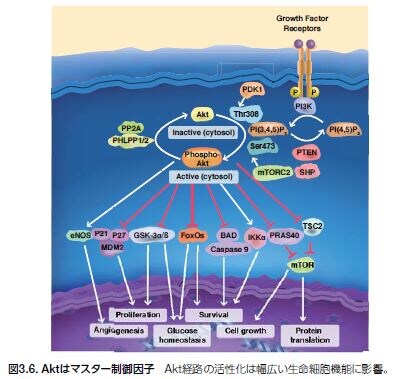 Aktは、Akt1/PKBα、Akt2/PKBβ、およびAkt3/PKBγの3種類のアイソフォームとして発現します。3種類のアイソフォームには全てに、アミノ末端プレクストリン相同(PH)ドメイン、中央のセリン/スレオニン触媒ドメイン、および小さなカルボキシル末端調節ドメインが存在します。PHドメインは、PI3K産物であるPIP2およびPIP3に結合します(図3.7)*18,19。これらの相互作用によりAktの細胞膜への移行が誘導され、ホスホイノシチド依存性キナーゼ1(PDK1)によって触媒ドメインの活性化ループ中のThr308がリン酸化されます *20 。
Aktは、Akt1/PKBα、Akt2/PKBβ、およびAkt3/PKBγの3種類のアイソフォームとして発現します。3種類のアイソフォームには全てに、アミノ末端プレクストリン相同(PH)ドメイン、中央のセリン/スレオニン触媒ドメイン、および小さなカルボキシル末端調節ドメインが存在します。PHドメインは、PI3K産物であるPIP2およびPIP3に結合します(図3.7)*18,19。これらの相互作用によりAktの細胞膜への移行が誘導され、ホスホイノシチド依存性キナーゼ1(PDK1)によって触媒ドメインの活性化ループ中のThr308がリン酸化されます *20 。
完全な活性化には、第二の部位Ser473のリン酸化反応が必要とされます。現在の研究では、mTOR–Rictor複合体(mTORC2)が第二の部位でのリン酸化反応に関与する主要なキナーゼであることが示唆されていますが、Akt活性化にはインテグリン結合キナーゼ(Ilk)PDK1、DNA依存性プロテインキナーゼ(DNA-PK)、および血管拡張性失調症変異(ATM)といった他のキナーゼも関与していると考えられています *21 。
がんにおけるAktの役割
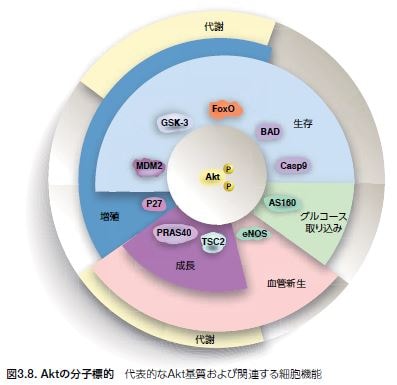
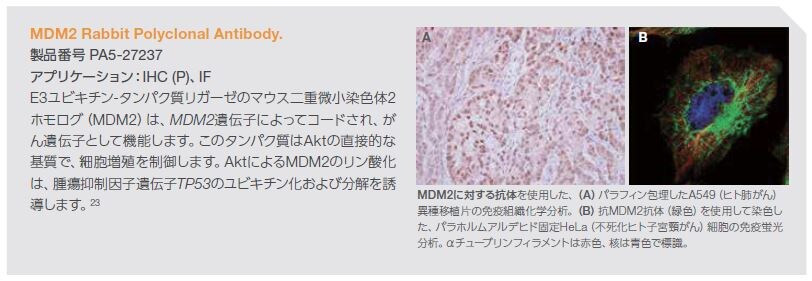
PTENの機能不全はAktの制御不能な活性化に密接に関係しており、腫瘍細胞増殖につながります。活性化されたAktは、アポトーシス、遺伝子転写、細胞周期進行、および細胞代謝の制御因子を含む腫瘍の発生に関連するプロセスを制御する下流のシグナル伝達基質を標的とします。Aktの基質となる分子は50種類以上知られており、細胞増殖の制御に関与する直接的なAkt基質にはMDM2、p21、およびp27が含まれます(図3.8)*22 。
Aktアイソフォームに影響する増殖および活性化変異は、複数のタイプのヒトがんにおいて同定されてきました。そのため、Aktシグナル伝達経路、ならびに下流経路における調節不全は多くのがんのホールマークの1つであり、Aktは抗がん剤および治療法の開発のための重要な標的となっています *3。
Akt変異を持つヒトがん
- 髄膜
- 乳房
- 子宮内膜
- 尿路
- 甲状腺
- 皮膚
- 肺
- 卵巣
- 造血器およびリンパ系
- 腎臓
哺乳類ラパマイシン標的タンパク質(mTOR)
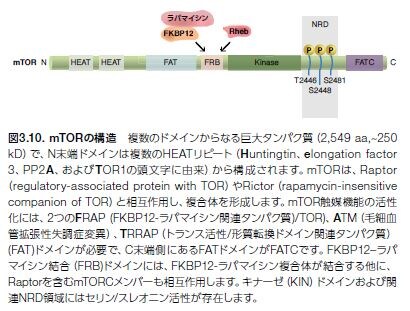
mTORの構造、機能および調節
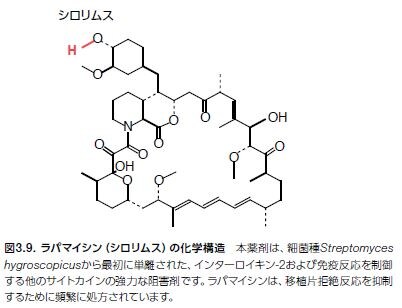
ラパマイシン̶抗真菌性抗生物質でTリンパ球活性の強力な阻害剤̶に関する研究は、mTORの発見およびシグナル伝達経路の解明につながります *24–26(図3.9および図3.10)。mTORは、PI3K関連プロテインキナーゼ(PIKK)ファミリーのメンバーで、血管拡張性失調症変異(ATM)、DNA依存性プロテインキナーゼ(DNAPK)および核酸の監視および修復プロセスに関連するいくつかの他のタンパク質が含まれます。mTORは、増殖因子経路のシグナルを伝播する機能を持ち、それによって細胞の成長、増殖、および生存をサポートします。さまざまながんにおいて、上方制御されているmTORシグナル伝達が検出されており、mTORを標的とする創薬努力が成功を収めています *27,28。 現在、進行性腎細胞がん、ならびに乳房、膵臓、および他の器官の特定の固形がんに対する治療薬として、2つのmTOR阻害剤が医薬品規制機関に承認されています(表3.2)。
mTORの機能および制御
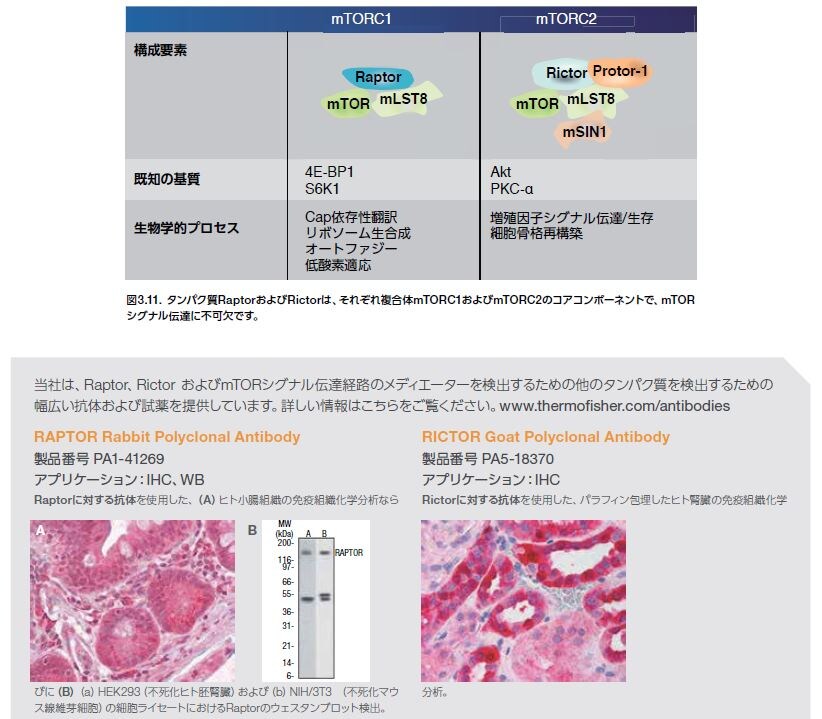
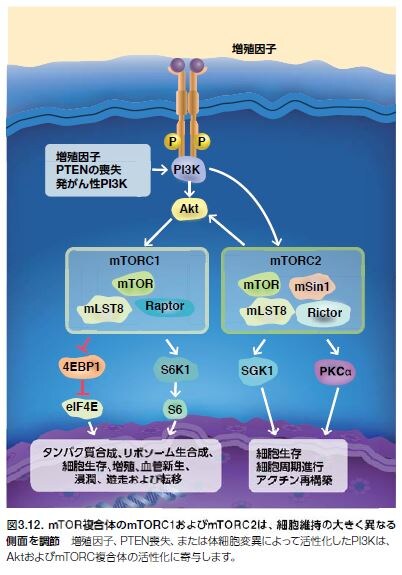
PI3K/Akt経路の下流エフェクターであるmTORは、複合体を形成し、複数のメカニズムによって活性化されます。mTORは、mTORシグナル伝達の種々の側面を制御するさまざまな調節タンパク質のサブユニットと複合体を形成します(図3.9および3.10)。mTORは、2種類のタンパク質複合体mTORC1およびmTORC2のコア触媒ユニットです。mTORC1複合体は、ラパマイシン感受性で、mTOR、Raptor、およびmLST8から構成されます。mTORC1は、翻訳抑制因子4E-BP1およびリボソームタンパク質S6キナーゼ(S6K)の両方をリン酸化することによって、細胞の成長および増殖を制御します。翻訳、リボソーム生合成、オートファジー、グル
コース代謝、低酸素に対する細胞反応、および転移などのさまざまな腫瘍細胞特異的なプロセスを含むその他の生物学的プロセスもmTORC1によって制御されます *30。 mTORC2複合体は、ラパマイシン抵抗性で、mTOR、Rictor、mLST8、およびmSin1から構成されます。mTORC2複合体は、複数のタンパク質̶S473上のセリンおよびスレオニンキナーゼAkt、血清グルココルチコイド調節キナーゼ1(SGK1)、ならびにプロテインキナーゼC α(PKCα)̶をリン酸化します。mTORC2は、細胞の生存および増殖を制御するために機能します(図3.12)*27。
これらのシグナル伝達経路について詳細に理解することによって、腫瘍進行に関連するキーメディエーターを標的として阻害する薬剤の合理的な医薬品開発が可能となりました*31 。例えば、VEGF、PDGFR、EGF、およびPI3K/Akt/mTOR経路を活性化する多くの他の経路を介するシグナル伝達を制限するチロシンキナーゼ受容体阻害剤が開発されてきました(表2.1)。同様に、mTORシグナル伝達を直接阻害する薬剤が腎細胞がんおよび他の固形腫瘍患者に対する治療薬として開発され、承認されています。
がんにおけるmTORの役割
mTORの阻害
増殖因子経路はmTORによって活性化される他に、低酸素状態およびアミノ酸の低レベル状態によって開始されます(図3.12)*32,33 。低酸素状態の持続は生命に関わる可能性があり、がん細胞は酸素正常状態を維持するための生存戦略に対応しています。転写因子の低酸素誘導因子1α(HIF-1α)は、血管新生の主要な制御因子で、低酸素に対する細胞反応に必要とされます。PI3K/Akt/mTOR経路は、HIF-1遺伝子発現レベルを増大させ、低酸素条件下での腫瘍細胞増殖に対する生存シグナルを伝達します。VHL遺伝子は、HIF-1αの分解に必要とされるフォンヒッペル・リンドウ腫瘍抑制因子をコードします。低酸素条件下において、HIF-1αはVEGF、PDGF、および血管新生を媒介する他のタンパク質を制御する遺伝子の転写を促進します。HIF-1α、mTOR、および関連する調節タンパク質の発現は、腫瘍細胞が低酸素状態に対応し、克服することを可能とします *34 。低酸素は血管新生の強力なインデューサーであるため、HIF-1αの制御因子であるTORは医薬品開発の標的となります。現在、2種類のmTOR阻害剤̶いずれもapalogs(ラパマイシンのアナログ)̶が固形腫瘍に対する抗血管新生療法への使用について承認されています(表3.2)*28 。
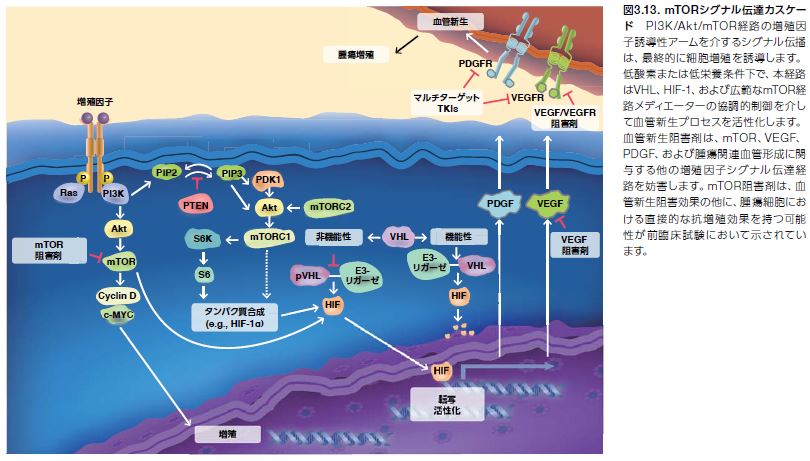 フォンヒッペル・リンドウ症候群は、VHL遺伝子における生殖細胞系列変異によって引き起こされる家族性がん症候群です。本症候群は、特定のタイプの悪性腫瘍および良性腫瘍を発現するリスクを増加させます。VHLの孤発性変異は、腎臓のさまざまながんにも関連します *37 。この遺伝子にコードされるタンパク質は、ユビキチンリガーゼE3活性を持ち、エロンギンB、エロンギンC、およびカリン-2のタンパク質を含む複合体を形成します。VHLタンパク質は、低酸素状態で生じる特定の細胞反応を制御する転写因子である低酸素誘導因子(HIF)のユビキチン化および分解に必要とされます *38 。
フォンヒッペル・リンドウ症候群は、VHL遺伝子における生殖細胞系列変異によって引き起こされる家族性がん症候群です。本症候群は、特定のタイプの悪性腫瘍および良性腫瘍を発現するリスクを増加させます。VHLの孤発性変異は、腎臓のさまざまながんにも関連します *37 。この遺伝子にコードされるタンパク質は、ユビキチンリガーゼE3活性を持ち、エロンギンB、エロンギンC、およびカリン-2のタンパク質を含む複合体を形成します。VHLタンパク質は、低酸素状態で生じる特定の細胞反応を制御する転写因子である低酸素誘導因子(HIF)のユビキチン化および分解に必要とされます *38 。
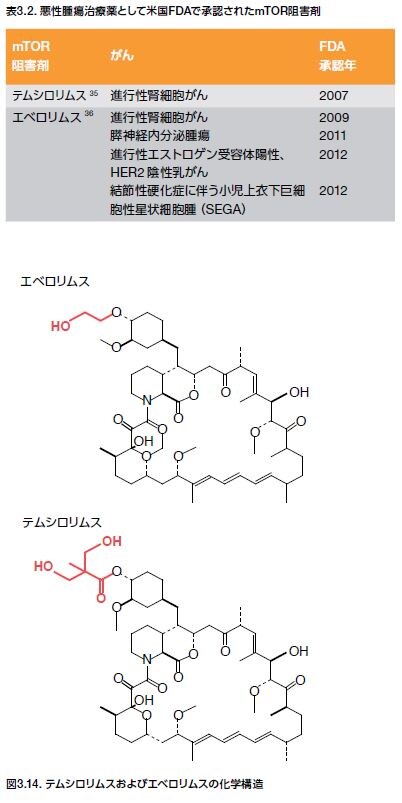
mTOR阻害剤ががん治療薬として現在承認されている部位の他にも、他の身体領域の組織においてmTORシグナル伝達に影響を及ぼす変異が検出されています *3 。
mTOR変異を持つヒトがん
- 子宮内膜
- 腎臓
- 結腸直腸
- 肺
- 皮膚
- 食道
- 上気道
- 尿路
- 乳房
- 卵巣
- 造血器およびリンパ系
- 肝臓
- 膵臓
- 脳
PI3K/Akt/mTOR調節と他の経路間のクロストークを制御するプロセスを解明することによって、医薬品規制当局に承認される、さまざまながんを治療標的としたmTOR阻害剤を合理的に開発することが可能となりました。第一世代のmTOR阻害剤は、特定の条件下で有効性を示しますが、主としてmTORC1媒介性プロセスを阻害します。両方のmTORC複合体を標的とする第二世代の阻害剤は、開発中で、 腫瘍誘導性Akt/mTORプロセスをブロックする効果がより高いと考えられます *39 。
当社は、mTOR経路を検出するための幅広い抗体および試薬を提供しています。詳しい情報はこちらをご覧ください。

【無料ダウンロード】がん増殖シグナル伝達経路の探索ガイドブック
当記事は、がん増殖シグナル伝達経路の探索ガイドブックからの抜粋です。下記の内容を含むPDFは下記から無料でダウンロード頂けます。
~主な内容~
- がん概論
- がん増殖シグナル伝達
- PI3K/Akt/mTORシグナル伝達経路
- Ras/Raf/MEK/ERK(MAPK)シグナル伝達経路
- Wnt/β-カテニン シグナル伝達経路
- 抗体:がん研究のための協力なツール
【無料ダウンロード】がん研究を促進させる遺伝子解析技術ハンドブック
また、複数のがん研究領域における興味深い最新の発見を可能にする遺伝子解析技術を紹介するハンドブックも配布しています。併せてごらんください。
参考文献:
1. Frumand DA, Rommel C (2014) PI3K and Cancer: lessons, challenges and opportunities. Nat Rev Drug Discov 13(2):140–156.
2. LoPiccolo J, Blumenthal GM, Bernstein WB, Dennis PA (2008) Targeting the PI3K/Akt/mTOR Pathway: effective combinations and clinical considerations. Drug Resist Updat 11(1–2):32–50.
3. Polivka J Jr, Janku F (2014) Molecular targets for cancer therapy in the PI3K/AKT/mTOR Pathway. Pharmacol Ther 142(2):164–175.
4. National Library of Medicine (US), Genetics Home Reference website (2012) Cowden syndrome. http://ghr.nlm.nih.gov/condition/cowden-syndrome
5. Stryer, L (1995) Biochemistry 4th ed, New York (NY): WH Freeman and Company. pp 265 and 344.
6. Rommel C, Camps M, Ji H (2007) PI3K delta and PI3K gamma: partners in crime in inflammation in rheumatoid arthritis and beyond? Nat Rev Immunol 7(3):191–201.
7. Grabon A, Khan D, Bankaitis VA (2015) Phosphatidylinositol transfer proteins and instructive regulation of lipid kinase biology. Biochim Biophys Acta 1851(6):724-735.
8. Koyasu S (2003) The role of PI3K in immune cells. Nat Immunol 4(4):313–319.
9. Vanhaesebroeck B, Stephens L, Hawkins P (2012) PI3K signaling: the path to discovery and understanding. Nat Rev Mol Cell Biol 13(3):195–203.
10. Dorsam RT, Gutkind SJ (2007) G-protein-coupled receptors and cancer. Nat Rev Cancer 7(2):79–94.
11. Jabbour E, Ottmann OG, Deininger M Hochhaus A (2014) Targeting the phosphoinositide 3-kinase pathway in hematologic malignancies. Haematologica 99(1):7–18.
12. Karakas B, Bachman KE, Park BH (2006) Mutation of the PIK3CA oncogene in human cancers. Br J Cancer 94(4):455–459.
13. Song MS, Salmena L, Pandolfi PP (2012) The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol 13(5):283–296.
14. Gu J, Tamura M, Pankov R et al. (1999) Shc and FAK differentially regulate cell motility and directionality modulated by PTEN. J Cell Biol 146(2):389–403.
15. National Library of Medicine (US), Genetics Home Reference website (2012) Bannayan-Riley-Ruvalcaba syndrome. http://ghr.nlm.nih.gov/condition/bannayan-riley-ruvalcaba-syndrome
16. Rozakis-Adcock M, McGlade J, Mbamalu G et al. (1992) Association of the Shc and Grb2/Sem5 SH2-containing proteins is implicated in the activation of the Ras pathway by tyrosine kinases. Nature 360(6405):689–692.
17. National Library of Medicine (US), National Center for Biotechnology Information website, gene database (2015) SHC1 SHC (Src homology 2 domain containing) transforming protein 1 [Homo sapiens (human)]. http://www.ncbi.nlm.nih.gov/gene/6464
18. Davies MA (2011) Regulation, role, and targeting of Akt in cancer. J Clin Oncol 29(35):4715–4717.
19. Arencibia JM, Pastor-Flores D, Bauer AF et al. (2013) AGC protein kinases: from structural mechanism of regulation to allosteric drug development for the treatment of human diseases. Biochim Biophys Acta 1834(7):1302–1321.
20. Song G, Ouyang G, Bao S (2005) The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med 9(1):59–71.
21. Riaz A, Zeller KS, Johansson S (2012) Receptor-specific mechanisms regulate phosphorylation of AKT at Ser473: role of RICTOR in 1 integrin-mediated cell survival. PLoS One 7(2):e32081.
22. Crowell JA, Steele VE, Fay JR (2007) Targeting the AKT protein kinase for cancer chemoprevention. Mol Cancer Ther 6(8):2139–2148.
23. Ogawara Y, Kishishita S, Obata T et al (2002) Akt enhance Mdm2-mediated ubiquitination and degradation of p53. J Biol Chem 277(24):21843–21850.
24. Vézina C, Kudelski A, Sehgal SN (1975) Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J Antibiot (Tokyo) 28(10):721–726.
25. Sehgal SN (2003) Sirolimus: its discovery, biological properties, and mechanisms of action. Transplant Proc 35(3 Suppl):7S–14S.
26. Hoeffer CA, Klann E (2010) mTOR Signaling: at the crossroads of plasticity, memory, and disease. Trends Neurosci 33(2):67–75.
27. Ballou LM, Lin RZ (2008) Rapamycin and mTOR kinase inhibitors. J Chem Biol 1(1–4): 27–36.
28. Houghton PJ (2010) Everolimus. Clin Cancer Res 16(5):1368–1372.
29. Carracedo A, Pandolfi PP (2008) The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene 27(41):5527–5541.
30. Lu M, Wang J, Ives HE, Pearce D (2011) mSIN1 protein mediates SGK1 protein interaction with mTORC2 protein complex and is required for selective activation of the epithelial sodium channel. J Biol Chem 286(35):30647–30654.
31. Furman DA, Rommel C (2014) PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov 13(2):140–156.
32. Serova M, de Gramont A, Tijeras-Raballand A et al. (2013) Benchmarking effects of mTOR, PI3K, and dual PI3K/mTOR inhibitors in hepatocellular and renal cell carcinoma models developing resistance to sunitinib and sorafenib. Cancer Chemother Pharmacol 71(5):1297–1307.
33. Nishikawa T, Takaoka M, Ohara T et al. (2013) Antiproliferative effect of a novel mTOR inhibitor temsirolimus contributes to the prolonged survival of orthotopic esophageal cancer-bearing mice. Cancer Biol Ther 14(3):230–236.
34. Harris AL (2002) Hypoxia—a key regulatory factor in tumour growth. Nat Rev Cancer 2(1):38–47.
35. Pfizer Laboratories (2014) Torisel (temsirolimus) prescribing information. http://labeling.pfizer.com/showlabeling.aspx?id=490
36. Novartis (2015) Afinitor (everolimus) prescribing information. https://www.pharma.us.novartis.com/product/pi/pdf/afinitor.pdf
37. Neumann HP, Zbar B (1997) Renal cysts, renal cancer and von Hippel-Lindau disease. Kidney Int 51(1):16–26.
38. Ivan M, Kondo K, Yang H et al. (2001) HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292(5516):464–468.
39. Zhang YJ, Duan Y, Zheng XF (2011) Targeting the mTOR kinase
研究用にのみ使用できます。診断目的およびその手続き上での使用はできません。
記事へのご意見・ご感想お待ちしています

予測ゲノミクスでがんを理解する
この記事は、科学者向けにがん研究の新しい...
Read More
がん研究を促進するがんオルガノイド(Tumoroid)の培養・解析製品まとめ
がんオルガノイド(Cancer organoid、Tumoroid)は�...
Read More
リキッドバイオプシーを活用した肺がんと悪性脳腫瘍に対する研究事例のご紹介
リキッドバイオプシーは血液や尿などの体液...
Read More
オルガノイド実験に使える定番製品まとめ(ヒト多能性幹細胞用の培地と基質編)
オルガノイドは現在の生命科学でもっともホ...
Read More