▼もくじ [非表示]
神経変性疾患概論
神経変性とは、細胞の構造および機能の損失をきたしているニューロンを指す一般的用語です *1 。神経変性疾患の最も一般的な例としては、アルツハイマー病(AD)、パーキンソン病(PD)、ハンチントン病、および筋委縮性側索硬化症(ALS)が挙げられます。神経変性疾患(ND)は消耗性疾患で、全世界の罹患者数は急増し続けています *2,3 。最新の治療法は、疾患の初期段階に破壊される特定のニューロンおよびシグナル伝達系をターゲットとしています。その例として、ADにおいてレベルが上昇するアセチルコリンに対するアセチルコリンエステラーゼ阻害剤や、PDに対するドーパミン作動薬があります。現在の治療法は、よくても症状を軽減するだけで、根本的な病状に対処したり、疾患経過を有意に緩和あるいは遅延させることはありません。そのため、神経の保護および再生につながる新しい治療法の開発が急務とされています。例えば、ヒト胚性幹細胞(ESC)や人工多能性細胞(iPSC)をベースとするような細胞置換療法が、様々なNDの治療を成功させる可能性があります。また、神経炎症は神経変性障害の生理病理学に基本的に関与しているため、神経炎症反応を調節する戦略が新しい治療の選択肢となる可能性があります *3,4 。
本記事は、神経変性疾患概論を提供し、新規および新興の技術ならびにさらなる神経科学研究を可能とする抗体ベースのツールに関する情報を紹介することを目的としています。
※本記事の末尾において、神経変性疾患について体系的に学べる無料コンテンツを紹介しています。
一般統計
全世界で数百万人が何らかのNDに罹患しています。5 ADおよびPDの罹患率が最も高く、米国ではそれぞれ500万人以上および最大100万人が罹患しています(表1.1)。何らかの神経変性疾患を持つ患者の数は上昇し続けています。例えば、65歳以上のAD罹患者は、2050年までに3倍になることが予測されています *6-16 。
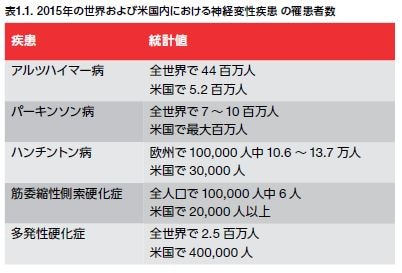
リスクファクター
遺伝因子は、早期発症型、家族型および孤発型の神経系を侵す変性疾患に起因します。継承された単一遺伝子変異は優性または劣性形式で発現し、家族性NDの疾患発症の予測は可能であるのに対し、他の遺伝子変異では孤発性疾患が発現する傾向があります。例えば、α-シヌクレインタンパク質をコードする遺伝子であるシヌクレイン,α(アミロイド前駆体の非A4成分)(SNCA)における変異はPDに関与している一方、アミロイドβ(A4)前駆体タンパク質(APP)遺伝子における多様な障害はADの様々な形態と関係しています。様々なNDに関連する遺伝子の代表例を表1.2に示します。複数の様々な遺伝的および環境的リスクファクターがNDの発症に関係しています(図1.1)。さらに、加齢も多くのNDの発症に関係する別のリスクファクターであり、恐らく環境因子への曝露の蓄積や細胞修復機構の機能低下によるものと考えられます。心血管疾患や頭部外傷による神経損傷などの症状も、AD発症リスクを上昇させます。一方、喫煙および特定の感染性病原体はMSのリスクを上昇させます *17-20。
抗体:神経変性疾患研究のための強力なツール
抗体は、免疫蛍光イメージング、免疫組織化学、ウェスタンブロッティング、ELISA、フローサイトメトリー、および他のアプリケーションを含む複数のアプリケーションで使用されています。当社の抗体が優れた実験結果の達成を可能とすることは世界中の多くの文献で実証されています。抗体検索ツールにアクセスし、目的の研究に適した抗体を見つけるためにはこちらをご覧ください。
参考文献:
1. Przedborski S, Vila M, Jackson-Lewis V (2003) Neurodegeneration: what is it and where are we? J Clin Invest 111(1):3–10.
2. Davis M, Stroud C, eds (2013) Neurodegeneration: Exploring Commonalities Across Diseases: Workshop Summary USA: National Academy of Sciences. p4-15. http://www.nap.edu/catalog.php?record_id=18341. Accessed September 2015.
3. Aoun S, McConigley R, Abernethy A Currow DC et al. (2010) Caregivers of people with neurodegenerative diseases: profile and unmet needs from a population-based survey in South Australia. J Palliat Med 13(6):653–661.
4. Amor S, Puentes F, Baker D et al. (2010) Inflammation in neurodegenerative diseases. Immunology 129(2):154–169.
5. Neurodegenerative Diseases. National Institute of Environmental Health Sciences. https://www.niehs.nih.gov/research/supported/diseases/neurodegenerative/index.cfm. Accessed July 2015.
6. Alzheimers.net 2015 Alzheimer’s Statistics. http://www.alzheimers.net/resources/alzheimers-statistics/. Accessed July 2015.
7. Statistics on Parkinson’s. Parkinson’s Diseae Foundation http://www.pdf.org/en/parkinson_statistics Accessed July 2015.
8. Bates GP, Dorsey R, Gusella J, et al. (2015) Huntington Disease. Nat Rev Dis Primers 1:1–21.
9. Kinsley L, Siddique (2001) T Amyotrophic Lateral Sclerosis Overview. [Updated 2015 Feb 12]. In: GeneReviews™ [Internet]. Pagon RA, Adam MP, Ardinger HH etal., editors. Seattle (WA): University of Washington, Seattle; 1993–2015.
10. Epidemiology of ALS and Suspected Clusters. ALS Association. http://www.alsa.org/als-care/resources/publications-videos/factsheets/epidemiology.html. Accessed July 2015.
11. Epidemiology of MS. MS International Federation. http://www.msif.org/research/epidemiology-of-ms/. Accessed July 2015.
12. Tullman MJ (2013) Overview of the epidemiology, diagnosis, and disease progression associated with multiple sclerosis. Am J Manag Care 2013;19(2 Suppl):S15–S20.
13. Facts You Should Know. ALS Association. http://www.alsa.org/about-als/facts-youshould-know.html. Accessed July 2015.
14. What is ALS/MND? International Alliance of ALS/MND Associations. http://www.alsmndalliance.org/what-is-alsmnd/. Accessed July 2015.
15. Huntington’s Disease. Remedy’s Health Communities.com. http://www.healthcommunities.com/huntingtons-disease/overview-of-huntingtons.shtml. Accessed July 2015.
16. 2015 Alzheimer’s Disease Facts and Figures. Alzheimer’s Association. https://www.alz.org/facts/downloads/facts_figures_2015.pdf. Accessed July 2015.
17. Hindle JV (2010) Ageing, neurodegeneration and Parkinson’s disease. Age Ageing 39(2):156–161.
18. Risk Factors. Alz.org http://www.alz.org/alzheimers_disease_causes_risk_factors.asp. Accessed July 2015.
19. Simon KC, Schmidt H, Loud S (2015) Risk factors for multiple sclerosis,neuromyelitis optica and transverse myelitis. Mult Scler 21(6):703–709.
20. Van Deerlin VM (2012) The genetics and neuropathology of neurodegenerative disorders: perspectives and implications for research and clinical practice. ActaNeuropathol 124(3):297–303.
21. Klein C, Westenberger (2010) A Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med 2(1):a008888.
神経変性疾患における共通のテーマ
これまで、それぞれの神経変性疾患(ND)は、病因や病態の異なる個別のエンティティとして研究されてきました。しかしながら、近年、特定の分子、細胞、および遺伝的メカニズムがNDの多様性を広げている可能性があることが明らかとなってきています。これらの調査結果は、共通性に関する調査が基本的な病態プロセスに関する見識だけでなく、共通の診断バイオマーカーおよび治療ターゲットに関する見識も提供できる可能性があることを示唆しています *1 。
様々な脳領域にわたる類似した神経機能障害の特徴に加えて、NDは特定の病態生理学プロセスを共有していると考えられます。例えば、これらの疾患の多くは、不溶性プラークの出現を導くミスフォールドしたタンパク質の産生、蓄積、および排出の減少が関係しています。また、神経機能障害および破壊の一般的なメカニズムは、複数の神経変性疾患において生じており、炎症、酸化ストレス、ミトコンドリア機能不全、および興奮毒性増強として兆候が現れます(表2.1) *2-12 。
数十年の研究によって現在の治療法がもたらされていますが、多くのケースで、これらの疾患の経過において破壊される特定のニューロンおよびシグナル伝達系がターゲットとされています。これらの療法には、アルツハイマー病(AD)においてレベルが上昇するアセチルコリンに対するアセチルコリンエステラーゼ阻害剤やパーキンソン病(PD)に対するドーパミン作動薬が含まれます。既存の療法は、よくても症状を軽減あるいは遅延させるだけで、根本的な病状に対処したり、疾患経過を有意に緩和あるいは遅延させることはありません。また、既存の療法では神経機能を保存または修復することはできません。これに対し、開発中の新しい療法は神経破壊に関与する特定の病理過程をターゲットとしてデザインされており、神経を保護および修復することが可能です。例えば、間葉系幹細胞や人工多能性幹細胞(iPSC)由来細胞を用いる幹細胞ベースの療法は、様々なNDを治療するための次世代の治療法をリードするものと考えられます。
神経変性疾患概論
それぞれのNDには、疾患を決定付ける様々な特徴があります。例えば、それぞれのNDの病態形成には異なる脳領域が関与し、特有の細胞タイプが関与していると考えられます。例えば、ADではコリン作動性ニューロンが関与し、PDにはドーパミン作動性ニューロンの分解が関係し、そしてMSではCNSのニューロンの周りに存在するミエリン鞘を形成するオリゴデンドロサイトが影響を受けます。NDは、疾患特異的な特徴を持つにも関わらず、タンパク質のミスフォールディング、慢性炎症、および細胞死を誘導する高レベルな酸化ストレスなどの多くの共通する病状を共有しています。複数の神経変性疾患に関連する特徴の概要を表2.1に示します *13–20。
バイオマーカーおよびタンパク質を評価するための方法の選択
バイオマーカーの発見は、NDの初期段階での検出やこれらの消耗性症状を治療するための新しい療法の開発につながる戦略を発展させるのに不可欠です。また、細胞内におけるタンパク質の局在性および分布パターンを決定することによって、病態形成を促進するメカニズムに関する見識がもたらされます。数多くのアプローチにおいて、特定のNDに関連するバイオマーカーやタンパク質の細胞内局在性を明らかにするためのバイオマーカーが使用されています。物理的バイオマーカーは、脳イメージング法に分類されると考えられます。血液および脳脊髄液(CSF)に存在するタンパク質を同定および定量するための技術の例としては、ELISAやLuminexなどの様々なプロテオミクスプラットフォームが挙げられます *21。
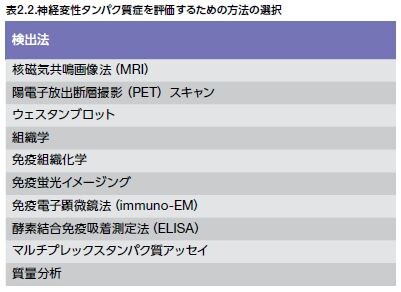
ニューロン集団を同定するための方法の例としては、アミノ酸、ペプチド、およびタンパク質の検出を可能とする免疫電子顕微鏡法(immuno-EM)の使用が増加してきています。この手法には、特別な組織調製プロトコルおよび金コロイド粒子のような高電子密度マーカーが必要とされます(表 2.2および図2.1)。
病変を直接可視化するためにヒト由来CNS組織サンプルを取得することには大きな制限があります。しかしながら、ADおよび他のND関連タンパク質の組織学的および免疫組織化学分析を用いる死後組織分析は診断補助に一般的に使用されており、初代神経細胞および細胞株を使用するin vitro細胞実験は抗体ベースのツールを用いる実験にもよく用いられています *22 。これらのツールは、多くの場合、CNSなどのサロゲートコンパートメント試験用、ならびに非ヒト霊長類およびげっ歯類を含む幅広い様々な生物種に発現させた神経変性モデルの特性決定を行うための実験エンドポイントとして頻繁に使用される神経組織解析用として使用されます。前臨床試験モデルを使用する際の大きな課題は、ヒトNDの複雑さを完全に再現できないことです。しかしながら、実験モデルを使用することは、バイオマーカーを同定したり、薬剤、および神経変性状態を治療するためにデザインされた細胞ベースの治療法についてテストする際の助けとなり、NDの発病および進行に関与する細胞および分子メカニズムに関する見識を提供することにつながります *23 。
当記事は、神経変性疾患ガイドブックからの抜粋です。
下記の内容を含むPDFは下記から無料でダウンロード頂けます。
タンパク質のミスフォールディングおよび神経変性
タンパク質ミスフォールディングのメカニズム
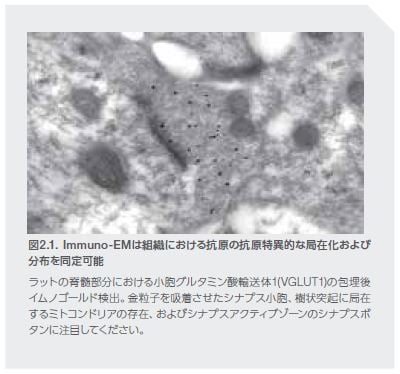
タンパク質のミスフォールディングは、AD、PD、HD、および筋委縮性側索硬化症(ALS)を含む多くの神経変性疾患の顕著な特徴です。ミスフォールドしたタンパク質は、一連の小胞体(ER)ストレス反応を開始するシグナルを発生しますが、これは非折り畳みタンパク質応答(UPR)と総称され、有害なタンパク質の蓄積から細胞を保護するように働きます *12。 多くの野生型タンパク質はミスフォールディングしたり、凝集体を形成する能力がありますが、遺伝子変異の結果、アミノ酸配列が変化し、折り畳まれて凝集する傾向の高いタンパク質が生成することがあります。
例えば、ADでは、複数の様々なAPP変異によってオリゴマーの形成および線維形成に寄与する異常Aβペプチドの生成が引き起こされます(図2.2)。凝集の増加は、タンパク質産物の過剰産生、あるいは代謝、酸化、および炎症性のストレスイベントの増加にもつながる可能性があります。
小胞体(ER)内には、タンパク質のプロセシング、フォールディング、および輸送における品質コントロールのエレガントなメカニズムが存在し、ミスフォールドしたタンパク質の蓄積を回避しています。フォールダーゼおよび分子シャペロンは、このプロセスにおいて密接な役割を果たしています。ミスフォールドしたタンパク質は、ER内に保持されるか、あるいはオートファジー(自食作用)またはプロテアソーム依存性ER関連タンパク質経路によって分解されます *24,25。
膜通過ストレスセンサーおよび下流の転写因子はUPRを有します *26。 関連するストレスセンサーとして機能するER膜タンパク質の例としては、イノシトール要求性トランスメンブレンキナーゼ/エンドリボヌクレアーゼ 1(IRE1)、プロテインキナーゼ様真核生物翻訳開始因子2αキナーゼ(PERK)、および活性化転写因子-6(ATF6)が挙げられます *25 。活性化されると、これらのタンパク質は、タンパク質の生成速度、タンパク質フォールディングを助けるタンパク質の発現、タンパク質凝集の防止、ならびにERで産生したタンパク質の逆輸送の促進および分解を含む複数のプロセスを調節します(表2.3)*12。

プロテアソーム依存性の小胞体関連分解およびオートファジー
ミスフォールドしたタンパク質を除去するための主要なメカニズムは2種類存在します。1つの経路では、ERからサイトゾルへ逆輸送され、そこで異常タンパク質がユビキチン化され、プロテアソーム分解を受けます。これは小胞体関連分解(ERAD)と呼ばれます。もう1つのUPRにより刺激されるミスフォールドしたタンパク質を除去するための経路はオートファジーで、大きなタンパク質凝集体およびリポソーム内部のプロテアーゼによって分解されたオルガネラを除去する機能を持ちます *26,27 。これらのプロセスにおいていくつかの変化が生じると神経変性疾患におけるミスフォールドしたタンパク質の蓄積につながります。これらのプロセスの概要を表2.4に示します。

オートファジーのステップ
マクロオートファジーはオートファジーの主要な経路の1つです。これは様々なシグナル分子アセンブリが関与する段階的なプロセスで、ATGと呼ばれる一連の中心タンパク質が動員されます(図2.3)。28,29 ATGタンパク質はオートファージ―の開始、小胞の核形成、およびオートファゴソームの延長に関与します。このプロセスに関与する他のタンパク質を表2.5にリスト表示します。
- 誘発ーオートファジーの開始を活性化するためには、mTORを不活性化してULK1のリン酸化を低下させる ことが必要とされます。
- 小胞の核形成ーオートファゴソームの構築は、PI3K液胞タンパク質選別タンパク質VPS34 およびVPS15を含むPI3K複合体の活性化に関与し、Beclin-1(BECN1-調節オートファジータンパク質[AMBRA1]における活性化分子)、UV照射耐性関連遺伝子(UVRAG)、およびBax-相互作用因子1(BIF1)とのコラボレーションにより生じます。
- 小胞の延長ーユビキノン様抱合系が小胞の延長を調節します。
- 小胞の回復ー小胞複合体内の大部分のタンパク質は小胞と結合した状態で留まらず、オートファジープロセスを介してリサイクルされます。
- 完了ーATGタンパク質は放出され、リサイクルされます。
- ドッキングおよび融合ーオートファゴソームはリソソームと融合し、オートリソソームを形成します。
- 小胞の崩壊および分解ーリソソーム酵素によりカーゴが分解されます。


アミロイドβタンパク質プラーク、タウタンパク質およびα-シヌクレイン封入体
最も蔓延している神経変性疾患は、神経の構造および機能を損傷するオリゴマーおよびミスフォールドしたタンパク質の不溶性の大きな凝集体が蓄積するという共通の特徴を共有します。これらの凝集体は、アルツハイマー病では、細胞外でアミロイドβ(Aβ)タンパク質プラーク、細胞内では高リン酸化タウが蓄積した神経原線維として、そしてα-シヌクレインが含まれるレビー小体として(最も代表的なのはパーキンソン病への関与)形成され存在します。これらの各タンパク質凝集体は、当初は際立ったNDの特徴として認識されましたが、現在では考慮すべきクロスオーバーがあると認識されています。例えば、Aβタンパク質プラークおよびα-シヌクレインはともに、PDと診断された患者の脳において高頻度で同定されます。リン酸化タウタンパク質の異常な凝集体は、ADに関連する神経毒性に寄与しますが、タウオパチーに分類される他の疾患例としてはこれらに限定されませんが、レビー小体型認知症および前頭側頭認知症が挙げられます。神経変性疾患に関連するタンパク質成分および他の特性に関する概要を表2.1に示します *30,31。
病的封入体におけるα-シヌクレインの存在はPDの特徴を定義付けるものですが、蓄積するこのタンパク質は、総称してシヌクレイン病と呼ばれる幅広い神経変性疾患に特徴的な病変の主要な非アミロイド成分です。これらの神経変性疾患には、これらに限定されませんが、パーキンソン病、レビー小体型認知症、ならびに家族性および孤発性アルツハイマー病、ダウン症、進行性自律神経不全症が含まれます *22。
 Aβ、タウおよびα-シヌクレイン検出用の抗体、ELISAおよびLuminexキットに関する詳しい情報についてはこちらをご覧ください。
Aβ、タウおよびα-シヌクレイン検出用の抗体、ELISAおよびLuminexキットに関する詳しい情報についてはこちらをご覧ください。
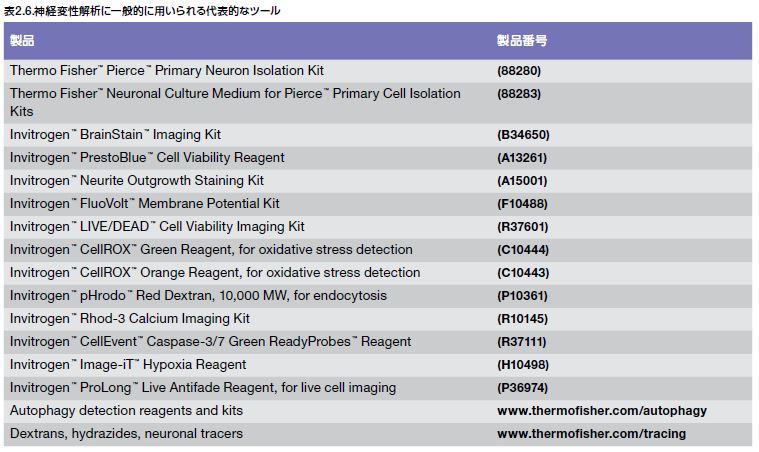
当社では、Thermo Fisher Scientificブランドの神経特異的ターゲット研究用の抗体、ELISA、およびLuminexアッセイの幅広い製品群に加えて、細胞生存率および増殖アッセイキット、神経解剖および機能分析用蛍光プローブ、ニューロントレーサー、神経変性生物学研究関連一次抗体およびキットの幅広いセレクション、ならびに初代細胞および幹細胞研究用の神経細胞培養試薬を含む神経変性研究のための多様な試薬およびキットのコレクションを提供しています(表2.6)。全製品についてはこちらをご覧ください。
当記事は、神経変性疾患ガイドブックからの抜粋です。
下記の内容を含むPDFは下記から無料でダウンロード頂けます。
酸化ストレスおよび神経変性
ミトコンドリア機能不全
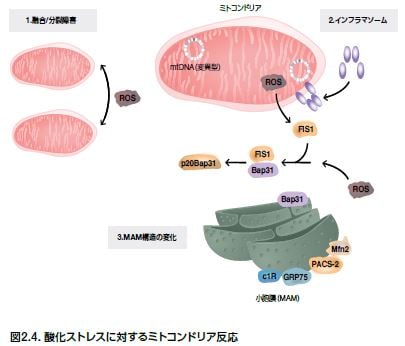
ミトコンドリア機能不全および酸化ストレスは、神経変性および加齢と密接な関係があり、様々なNDにおいて疾患特異的なミトコンドリア関連の変化が観察されます(図2.4および表2.7)*32-41 。例えば、ハンチンチンタンパク質およびポリQを含むフラグメントは、ミトコンドリア輸送障害および細胞内Ca2+濃度上昇を誘発します *42 。
PDでは電子伝達が低下し、AD、PDおよびALSではミトコンドリアDNA(mtDNA)における変異が検出されています *43 。活性酸素種(ROS)はミトコンドリア機能不全を促進し、アポトーシスを引き起こす可能性があります。ミトコンドリアの分裂と融合は、ミトコンドリアの健全性に必要とされる生理的プロセスですが、これらのプロセスの撹乱は、がん、心血管疾患、神経変性状態を含む数多くのヒト疾患において引き起こされます *44 。ミトコンドリアおよびER機能間のクロストークの複雑さについては完全には解明されていませんが、ミトコンドリアと小胞体の接触領域(MAM)が2つのオルガネラ間における脂質の移動およびCa2+のホメオスタシスを可能としており、オートファゴソーム形成の部位であることが研究において示されています。
インフラマソーム複合体も、ミトコンドリア機能不全に関与し、ピロトーシスを誘発します。これらの多タンパク質オリゴマーは、インターロイキン(1βおよび18)の産生を誘導し、プログラム細胞死に関与するカスパーゼを活性化する病原シグナルに反応して形成されます。NOD-様受容体ファミリータンパク質のメンバーであるNLRP1、3、および4は、ROSの生成を導くインフラマソームのキーコンポーネントです。これらのプロセスに関与するタンパク質の代表例を表2.8に示します *32-42。
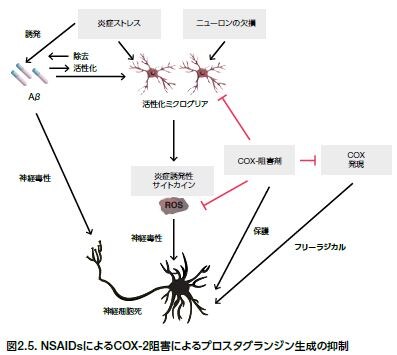
神経変性におけるCOX-2の役割
過剰なROSレベルは、サイトカイン、ペプチド、および病原構造などの複数の炎症メディエーターの産生を促進します。これらの要素は脳損傷に寄与し、シクロオキシゲナーゼ-2(COX-2)のアップレギュレーションの一因となります。プロスタグランジンの生成に必要とされるCOX-2は、様々なNDに関連する炎症に関与します(図2.5)。COX-2や炎症を阻害する薬理作用のある薬剤(非ステロイド性抗炎症薬[NSAIDs]など)は、AD、PD、ALS、およびMSなどの疾患進行の抑制あるいは軽減の目的で使用するために研究されています*45。

 ミトコンドリアタンパク質検出用抗体に関する詳しい情報についてはこちらをご覧ください。
ミトコンドリアタンパク質検出用抗体に関する詳しい情報についてはこちらをご覧ください。
神経変性疾患研究の新規分野
炎症
中枢神経系(CNS)は、免疫学的に特有の“免疫特権”部位です。インタクトな血液脳関門(BBB)は、末梢免疫細胞からCNSへの流入を制限します。ミクログリア活性化およびそれに続く免疫応答性を軽減する可溶性因子、ならびに神経抗原のリンパ排液は、免疫反応を誘発するよりもむしろ免疫寛容 (自己と非自己の区別の学習)を促進するように作用します。これらの防御機構にも関わらず、免疫系の自然免疫および適応免疫はともに様々なNDにおける神経損傷の開始および進行に影響を与えているとされます(表2.9)。脳における炎症誘発反応を抑制するための戦略の開発は、神経変性疾患治療の新しい選択肢につながります *46。

当社では、抗体に加えて、神経免疫学に関連するターゲットについて研究するための高度に検証された幅広いELISAおよびLuminexキットを提供しています。詳しい情報についてはこちらをご覧ください。
細胞ベースの療法の開発
幹細胞生物学、神経科学、およびリプログラミング技術の進歩は、研究の新しい道を切り拓きました。幹細胞は自己複製し分化細胞を産生するため、中枢および末梢神経系の障害および損傷のための幹細胞置換療法は現在不治のNDおよびニューロン損傷のための新しい治療法につながる可能性があります。正常な神経系の発達プロセスを再現するための戦略としては、内在性神経幹細胞の再生能力の活性化、あるいは胎児神経細胞または人工多能性幹細胞(iPSCs)由来細胞の移植が挙げられます。神経系における様々な最終分化細胞集団を生じさせる多能性幹細胞(PSCs)の分化経路を表2.6に示します。特定の神経生物学研究の目的をサポートするために、当社では複数のアプリケーションで検証済みの高特異的抗体の幅広いポートフォリオを提供しています。これらのツールは、グリア、タンパク質輸送、ニューロン新生、神経変性疾患、神経幹細胞研究、および他の神経生物学研究分野における研究を容易にします(表2.10)


【無料ダウンロード】神経変性疾患研究ガイドブック
当記事は、神経変性疾患研究ガイドブックからの抜粋です。以下の内容を含むPDFは下記から無料でダウンロード頂けます。
~主な内容~
- イントロダクション
- 神経変性疾患における共通のテーマ
- アルツハイマー病への取り組み
- 他の主要な神経変性疾患
- 抗体:神経変性疾患研究のための強力なツール
参考文献:
1. Davis M, Stroud C, eds (2013) Neurodegeneration: Exploring Commonalities Across Diseases: Workshop Summary USA: National Academy of Sciences. p4-15. http://www.nap.edu/catalog.php?record_id=18341. Accessed September 2015.
2. Prentice H, Pravinchandra JM, Wu JY (2015) Mechanisms of neuronal protection against excitotoxicity, endoplasmic reticulum stress, and mitochondrial dysfunction. Oxid Med Cell Longev Article ID 964518, in press.
3. Moussaud S, Jones DR, Moussaud-Lamodière, EL et al. (2014) et al. Alphasynuclein and tau: teammates in neurodegeneration? Mol Neurodegen. 9:43.
4. Trippier PC, Jansen Labby K, Hawker DD et al. (2013) Target- and mechanismbased therapeutics for neurodegenerative diseases: strength in numbers. J Med Chem 56(8):3121–3147.
5. Jin H, Kanthasamy A, Ghosh A et al. (2014) Mitochondria-targeted antioxidants for treatment of Parkinson’s disease: preclinical and clinical outcomes. Biochim Biophys Acta 1842(8):1282–1294.
6. Rao VK, Carlson EA, Yan SS et al. (2014) Mitochondrial permeability transition pore is a potential drug target for neurodegeneration. Biochim Biophys Acta 1842(8):1267–1272.
7. Swomley AM, Förster S, Keeney JT et al. (2014) Abeta, oxidative stress in Alzheimer disease: evidence based on proteomics studies. Biochim et Biophys Acta. 2014; 1842(8):1248–1257.
8. Ciechanover A, Kwon YT (2015) Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med 47:e147.
9. Peggion C, Sorgato MC, Bertoli A (2014) Prions and prion-like pathogens in neurodegenerative disorders. Pathogens 3(1):149–163.
10. Nah J, Yuan J, Jung YK et al. (2015) Autophagy in neurodegenerative diseases: from mechanism to therapeutic approach. Mol Cells 38(5):381–389.
11. Kesidou E, Lagoudaki R, Touloumi O et al. (2013) Autophagy and neurodegenerative disorders. Neural Regen Res 8(24):2275–2283.
12. Rao RV, Bredesen DE (2004) Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr Opin Cell Biol 16(6):653–662.
13. Soto C (2003) Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci 4(1):49–60.
14. Tullman MJ (2013) Overview of the epidemiology, diagnosis, and disease progression associated with multiple sclerosis. Am J Manag Care 19(2 Suppl):S15–20.
15. Van der Walt A, Butzkeuven H, Kolbe S et al. (2010) Neuroprotection in multiple sclerosis: a therapeutic challenge for the next decade. Pharmacol Ther 126(1):82–93.
16. Wilkins A, Scolding N (2008) Protecting axons in multiple sclerosis. Mult Scler 14(8):1013–1025.
17. Querfurth HW, LaFerla FM (2010) Alzheimer’s disease. N Engl J Med 362(4):329–344.
18. Kumar A, Singh A, Ekavi AS (2015) A review on Alzheimer’s disease pathophysiology and its management: an update. Pharmacol Rep 67(2):195–203.
19. Wu GF, Alvarez E (2011) The Immunopathophysiology of multiple sclerosis. Neurol Clin 29(2):257–278.
20. Miller BR, Bezprozvanny I (2010) Corticostriatal circuit dysfunction in Huntington’s disease: intersection of glutamate, dopamine and calcium. Future Neurol 5(5):735–756.
21. Persson S, Havton LA (2009) Retrogradely transported fluorogold accumulates in lysosomes of neurons and is detectable ultrastructurally using postembedding immuno-gold methods. J Neurosci Methods 184(1):42–47.
22. Tarawneh R, Galvin JE (2009) Dementia With Lewy Bodies and Other Synucleinopathies. In: Weiner MF, Lipton AM, eds. Textbook of Alzheimer Disease and Other Dementias Washington DC: American Psychiatric Publishing, Inc. pp 195–217.
23. http://www.neurodegenerationresearch.eu/uploads/media/JPND_Exp_Models_Final_report_Jan_2014_-_DM.pdf
24. Irwin DJ, Lee VM, Trojanowski JQ (2013) Parkinson’s disease dementia: convergence of -synuclein, tau and amyloid- pathologies. Nat Rev Neurosci 14(9):626–636.
25. Wang S, Kaufman RJ (2012) The impact of the unfolded protein response on human disease. J Cell Biol. 2012 Jun 25;197(7):857–867.
26. Hetz C, Mollereau B (2014) Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci 15(4):233–249.
27. Glick D, Barth S, Macleod KF (2010) Autophagy: cellular and molecular mechanisms. J Pathol 22(1):3–12.
28. Füllgrabe J, Klionsky DJ, Joseph B (2014) The return of the nucleus: transcriptional and epigenetic control of autophagy. Nat Rev Mol Cell Biol 15(1):65–74.
29. Chen Y, Klionsky DJ (2011) The regulation of autophagy – unanswered questions. J Cell Sci 124(Pt 2):161–70.
30. Bachhuber T, Katzmarski N, McCarter JF et al (2015) Inhibition of amyloidplaque formation by -synuclein. Nat Med 21(7):802–807.
31. Lee G, Leugers CJ (2012) Tau and tauopathies. Prog Mol Biol Transl Sci 107:263–293.
32. Smirnova E, Griparic L, Shurland DL et al. (2001) Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell 12(8):2245–2256.36.
33. Lalier L, Cartron PF, Juin P et al. (2007) Bax activation and mitochondrial insertion during apoptosis. Apoptosis 12(5):887–896.
34. NCBI. FIS1 fission, mitochondrial 1 [ Homo sapiens (human) ]. http://www.ncbi.nlm.nih.gov/gene/51024. Accessed August, September 2015.
35. Coll RC, Robertson AA, Chae JJ et al. (2015) A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015 Mar;21(3):248–255.
36. Wakana Y, Takai S, Nakajima K et al. (2008) Bap31 Is an Itinerant protein that moves between the peripheral endoplasmic reticulum (ER) and a juxtanuclear compartment related to ER-associated degradation. Mol Biol Cell 19(5):1825–1836.
37. Phosphofurin Acidic Cluster Sorting Protein 2. GeneCards.org. Http://www.genecards.org/cgi-bin/carddisp.pl?gene=PACS2. Accessed August, September 2015.
38. Liu Y, Liu W, Song XD et al. (2005) Effect of GRP75/mthsp70/PBP74/mortalinoverexpression on intracellular ATP level, mitochondrial membrane potential and ROS accumulation following glucose deprivation in PC12 cells. Mol Cell Biochem 268(1–2):45–51.
39. MFN2 Gene. GeneCards.org. http://www.genecards.org/cgi-bin/carddisp.pl?gene=MFN2. Accessed August, September 2015.
40. Sebastián D, Hernández-Alvarez MI, Segalés J et al. (2012) Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. PNAS 109(14):5523–5528.
41. Hong J, Sha S, Zhou L et al. (2015) Sigma-1 receptor deficiency reduces MPTP-induced parkinsonism and death of dopaminergic neurons. Cell Death Dis 23;6:e1832.
42. Li XJ, Orr AL, Li S (2010) Impaired mitochondrial trafficking in Huntington’s disease. Biochim Biophys Acta 1802(1):62–65.
43. Cha MY, Kim DK, Mook-Jung I et al. (2015) The role of mitochondrial DNA mutation on neurodegenerative diseases. Exp Mol Med 47:e150.
44. Youle RJ, van der Bliek AM (2012) Mitochondrial fission, fusion, and stress Science 337(6098):1062–1065.
45. Figueiredo-Pereira ME, Rockwell P, Schmidt-Glenewinkel T et al. (2015) Neuroinflammation and J2 prostaglandins: linking impairment of the ubiquitinproteasome pathway and mitochondria to neurodegeneration. Front Mol Neurosci 13;7:104.
46. Amor S, Peferoen LA, Vogel DY et al. (2014) Inflammation in neurodegenerative diseases–an update. Immunology 142(2):151–66
研究用にのみ使用できます。診断目的およびその手続き上での使用はできません。
記事へのご意見・ご感想お待ちしています

【やってみた】熱変性の温度を下げた影響をリアルタイムPCRで確かめてみた
PCR反応における熱変性(denature)は、アニー�...
Read More
予測ゲノミクスでがんを理解する
この記事は、科学者向けにがん研究の新しい...
Read More
神経科学におけるフラグメント解析:複数の疑問を解決する柔軟なツール
脳は驚くべき器官です。さらに驚くべきこと...
Read More
細胞培養関連の学習リソースまとめ~トレーニング・セミナー・ハンドブックなど~
「細胞培養」はライフサイエンス研究におけ...
Read More