アルツハイマー病への取り組み
アルツハイマー病(AD)は、記憶および行動における進行性認知機能障害を特徴とします *1,2 。これは認知症の中で最も多くみられる形態で、発病は一般的に孤発性で、非常に多くの環境的および遺伝的リスクファクターが関与しています *3,4。
コリン作動性ニューロンの障害および大脳皮質における神経伝達の低下が認知機能の低下を引き起こします。本疾患はタンパク質プラーク(アミロイドの細胞外神経組織への沈着)、およびリン酸化したタウタンパク質から構成される細胞内神経原線維変化の存在によって特徴付けられます *5 。 臨床的および神経病理学的症状のエビデンスに先立って、嗅内皮質、海馬、後帯状回皮質、および中側頭回などの脳領域における遺伝子発現プロファイルおよび細胞機能に撹乱が観察され得ます *6 。特にADは、アミロイドβによって誘発されるミトコンドリア機能不全、重度の酸化ストレス、DNA損傷、アポトーシス、および脳内ホメオスタシスを破壊し、シナプス機能障害を引き起こす他の壊滅的なプロセスを特徴とします *7。
現在承認済みのAD治療薬は、一部のAD罹患者の症状を一時的に軽減することはありますが、根本的に疾患プロセスを変化させることはありません *8 。ADの早期発見および根本的な性質を変化させる治療の必要性は、バイオマーカーの発見、脳の生体イメージング、神経炎症、ならびに幹細胞ベースの研究ツールおよび有望な治療法などの基礎的および橋渡しとなるようなADの新規な研究分野に対する究明に拍車を掛けます *9,10。
※本記事の末尾において、神経変性疾患について体系的に学べる無料コンテンツを紹介しています。
一般統計
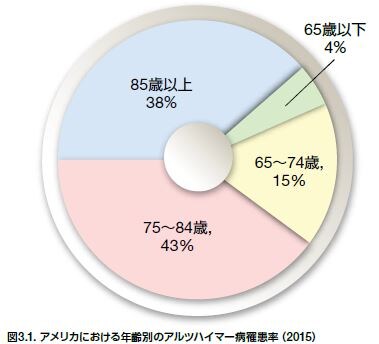
アルツハイマー病は神経変性疾患(ND)の最も一般的な形態で、65歳以上の人口の10%以上が罹患しているとされます *4 。アメリカでは約5.3百万人、世界ではほぼ44百万人がADを発症しています *4,11。 ADの発症およびAD発症のリスクは年齢の上昇とともに増加し、大部分のAD患者(>96%)は65歳を超えています(図3.1) *4 。疫学的評価において、アルツハイマー病は認知症の全症例の約60%~80%を占めています *12。
一般的な原因およびリスクファクター
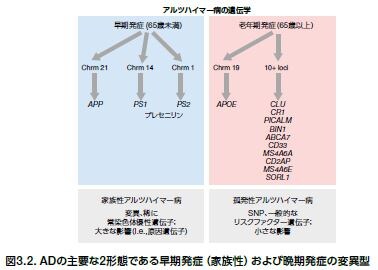
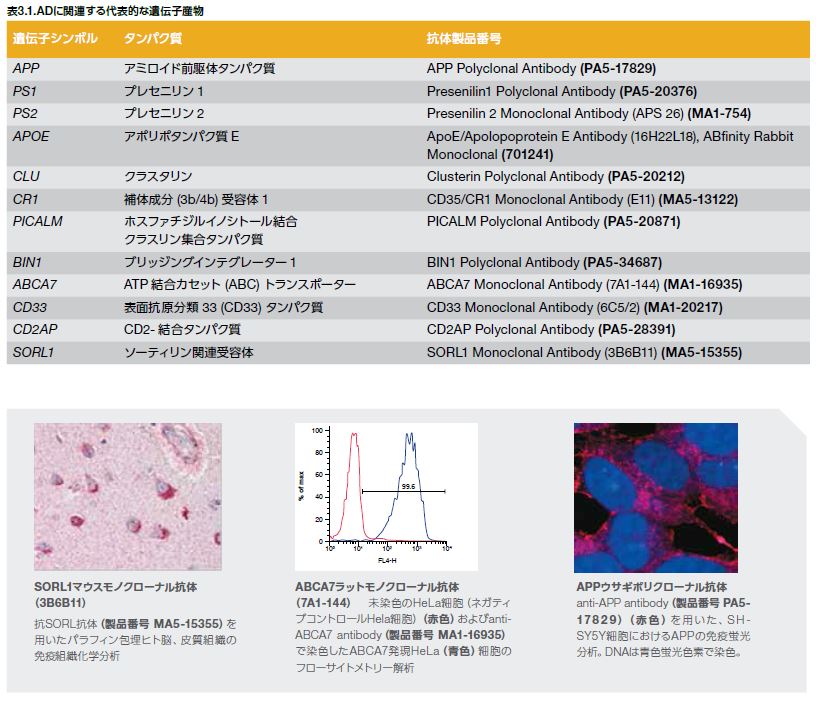
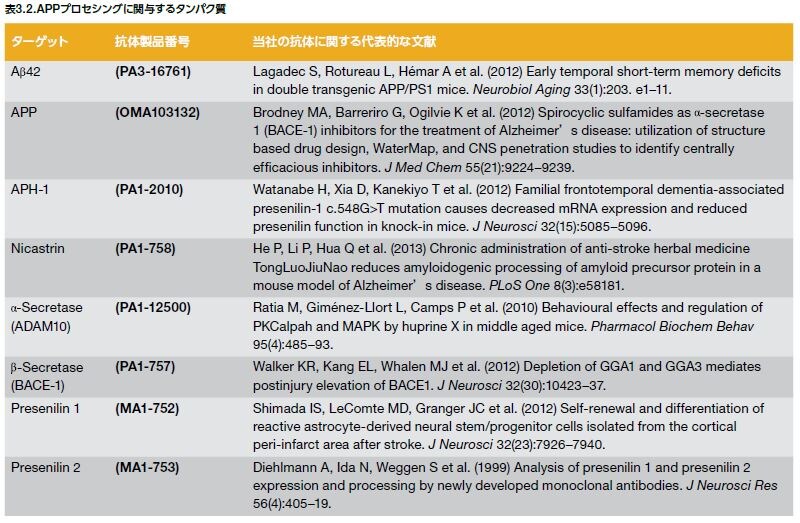
ADの家族歴は、個人のAD発症に影響する傾向があり、家族に1人以上罹患者がいる場合、発症リスクが上昇します。常染色体優性AD(ADAD)は、典型的には60歳以前に発症する早期発症型ADですが、AD症例の5%以下とされます。ADADに関与する遺伝子としては、アミロイド前駆体タンパク質(APP)およびプレセニリン-1および-2(PS-1およびPS-2)をコードする遺伝子が挙げられます。アポリポタンパク質E4(Apo E4)、および多くの他の遺伝子変異は、遅発性AD、あるいは孤発性AD(SAD)の発症リスクの上昇に関与するとされ、AD症例の大部分を占めています *13,14 。SADは遺伝的要因および環境要因の双方が関与して発症に至ると考えられています。ADに関与する遺伝子および遺伝子産物を図3.2および表3.1にリストアップします *15。
ADを発症させる一番のリスクファクターは加齢で、図3.2に示すような脳萎縮、炎症、フリーラジカルの生成、およびミトコンドリア機能不全などの変化を脳内に生じさせます。加齢は、ADの発症に関わる最大のリスクファクターですが、複数の環境要因および医学的状態によってリスクは増加します。高血圧、心疾患、脳卒中、糖尿病、および高コレステロール血症などの心血管障害は、ADの発症に関わる全身性炎症に寄与します。他の要因としては、座りがちな生活様式、肥満、口腔内の不衛生、および頭部外傷が挙げられ、特にそれが重度、反復的であったり、意識喪失を伴う場合はADの発症に寄与すると考えられます *13,16 。
病状
ADと診断された患者の約半数には、レビー小体型認知症(LBD)や血管性認知症などの他の認知症の形態に関連する病理学的特徴の痕跡がみられます *4 。ADの兆候は、老人斑や神経原線維変化などの神経病理学的変化が出現してから数年後に現れます。加齢に伴い、軽度の認知機能障害は現れる可能性がありますが、この中でADを発症するに至る患者は一部です *3 。疾患の進行は様々ですが、一般的には8~10年で死に至ります。治療法は存在しませんが、剖検前の診断は80~90%の確率で正確です *3 。この状況の厳しさから、早期の診断および疾患の進行速度を抑えるための治療オプションにつながる可能性のある新しい研究結果の発掘が重要であることは明らかです。
当記事は、神経変性疾患研究ガイドブックからの抜粋です。
ガイドブックは下記から無料でダウンロード頂けます。
アルツハイマー病の病理学および病態生理学
アルツハイマー病の特徴
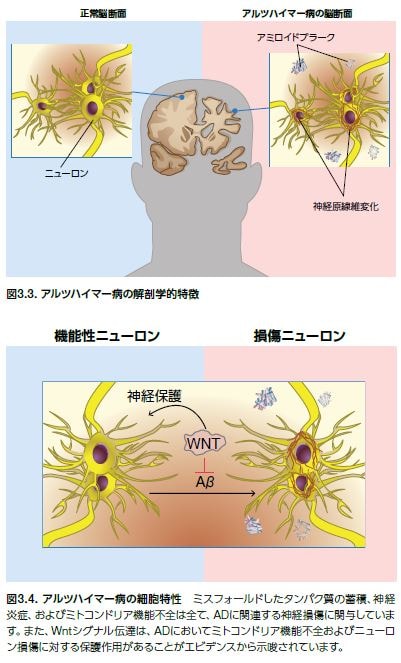
細胞外アミロイドプラークの存在、細胞内神経原線維変化、およびニューロン間のシナプス結合の喪失は、ADの顕微鏡レベルの主要な特徴です(図3.3)*17 。アミロイドプラークの存在は、臨床的に重要な記憶障害が発症する10~15年前に確認されます。AD患者の数は、α-シヌクレイン(レビー小体)および広範なエビデンスとして他のタンパク質の細胞内凝集の存在も示されます。実際、特に高齢患者において、ADとレビー小体型認知症、前頭側頭認知症、および脳血管疾患などの他のCNS疾患の間にはオーバーラップする症状があります*5 。肉眼によるADの解剖学的特徴としては、同年齢の健常者との比較における海馬の委縮および脳室の拡大が挙げられます。ADが進行するのに伴い、疾患後期に一時的で部分的な後頭葉および前頭部の委縮が生じます *18-20 。
ADの神経病理学的特徴としては、Aβ凝集物からなる細胞外プラークおよび高リン酸化タウの細胞内神経原線維変化が挙げられます。この壊滅的な疾患は、シナプスの損失および脳萎縮につながる神経細胞死によって特徴付けられます。Wntシグナル伝達経路は、神経機能およびシナプス維持の恒常性制御において極めて重要な役割を果たしています。しかしながら、このシグナル経路の撹乱はADの発症に関与しています。複数の研究において、Wnt3aリガンドを介するシグナル伝達はミトコンドリアの膜透過性を阻害することが示されています。同様に、Wnt5aリガンドシグナル伝達は、ADにおける核分裂核融合の変化を制限することによりミトコンドリアの完全性を保護します(図3.4)。
アミロイド前駆体タンパク質(APP)プロセシングの役割
アミロイド前駆体タンパク質(APP)は内在性膜タンパク質で、異なるプロセシングを受けて、様々なタンパク質断片を生成します。正常な状態では、APPは最初にα-セクレターゼによって切断され、sAPPαとC83というC末端断片を生じます。sAPPαの存在は、正常なシナプスシグナル伝達と関わっており、シナプス可塑性、学習と記憶、情動行動、およびニューロンの生存をもたらします。その後、C83がγ-セクレターゼによって分解され、アミロイド細胞内ドメインおよびp3を産生します。病的状態では、β部位アミロイド前駆体タンパク質–切断酵素(BACE-1)がAPPに作用し、可溶性APPβ (sAPPβ)を遊離します。これは細胞外空間に放出され、C99は細胞膜に保持されます(図3.5)。その後、C99はγ-セクレターゼで分解され、アミロイド細胞内ドメイン(AICD)を生じ、Aβが細胞外空間に放出されます。γ-セクレターゼは、プレセニリン1および2(PS-1およびPS-2)を含む複数のタンパク質から構成されます(表3.2)。PS-1およびPS-2をコードする遺伝子(PSEN1およびPSEN2)には、150を超える様々な変異が確認されていますが、大部分はPSEN1における変異です。PS変異はAPPプロセシングに影響を及ぼし、Aβ40よりもAβ42の凝集を増加させます *21,22 。
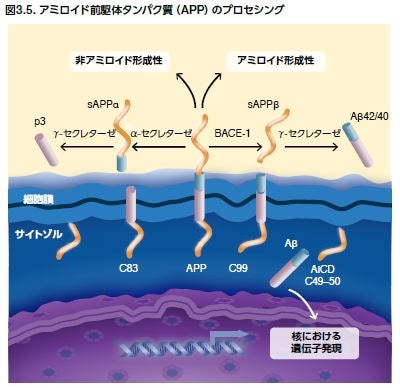
非アミロイド形成性アミロイド前駆体タンパク質(APP)のプロセシング中、α-セクレターゼはAPPに作用し、C83とsAPPαを産生します。sAPPαは正常なシナプスシグナル伝達に関わっています。その後、C83はγ-セクレターゼによって分解され、アミロイド細胞内ドメインおよびp3を産生します。アミロイド形成性APPのプロセシング中、β部位アミロイド前駆体タンパク質-切断酵素(BACE-1)はAPPに作用し、可溶性APPβ(sAPPβ)を遊離します。これは細胞外空間に放出され、C99は細胞膜に保持されます。γ-セクレターゼは、C99を切断してアミロイド細胞内ドメイン(AICD)を生じ、細胞外空間にAβを放出させる酵素複合体です。
抗体: 研究のための強力なツール
当社では、神経生物学関連の一次抗体およびイムノアッセイキットの幅広い製品群に加えて、神経解剖および機能分析用蛍光プローブ、ニューロントレーサー、ならびに初代細胞および幹細胞研究用の神経細胞培養試薬を含む神経科学研究を促進するための試薬を提供しています。
 当社の抗体ポートフォリオに関する詳しい情報についてはこちらをご覧ください。
当社の抗体ポートフォリオに関する詳しい情報についてはこちらをご覧ください。
アミロイドβおよびプラークの形成
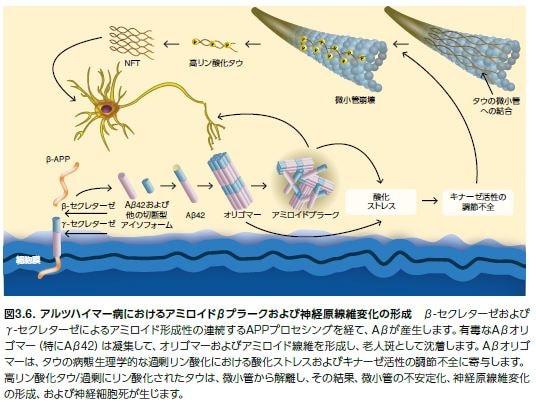
Aβペプチドは、正常な代謝において産生する、36~43アミノ酸からなる産物です。Aβ40モノマーは、自己凝集性が高い傾向のあるAβ42よりも高レベルで存在します。Aβ40とAβ42は高頻度で重合して、神経毒性を示すオリゴマーおよびより大きい凝集体を形成し、イオンチャンネルの遮断、カルシウム恒常性の崩壊、ミトコンドリア酸化ストレス、エネルギー代謝の障害、および最終的に神経細胞死につながる血糖調節異常を引き起こします。初期の研究ではアミロイドβの不溶性凝集体が有毒であることが示唆され、より最近のエビデンスでは可溶性オリゴマー前駆体も有毒であることが示唆されています *23,24 。また、認知機能障害は、全AβレベルよりもむしろAβオリゴマーのレベルに対応して発現します *22 。Aβの蓄積は、タウの過剰リン酸化および神経原線維変化の形成に先立って生じます(図3.6) *25。
タウタンパク質
微小管結合タンパク質(MAP)は、チューブリンに結合して、微小管の重合および安定性を促進し、リン酸化されたMAPを微小管から解離します *26 。タウは、ニューロンにおける主要なMAPで、正常な状態では、可溶性タンパク質として細胞体およびニューロンの軸索に位置します。ADにおいて、高リン酸化タウは、神経原線維変化(NFT)および神経絨毛糸(NT)の形成を導きます *27,28。
Aβの蓄積はADと関連しますが、正常な加齢(臨床的な認知症を除く)における脳内にも蓄積します。Aβは、ADの重症度との相関性よりも、軽度認知機能障害を有する患者のADに進行する可能性についての予測において重要であると考えられます。AD解析における新しいコンセプトでは、リン酸化タンパク質タウの病態生理学とADにおける臨床的認知症との高い相関関係に着目します *27,29 。このため、AD研究において、タウの役割にはより高い注目が集められています。
タウの特性
- 染色体17番上のMAPT遺伝子
- 別々のスプライシングによって産生する6種類の異なるタンパク質アイソフォーム
- 最大80のセリン/スレオニンリン酸化部位を含有
- 5つのチロシンリン酸化部位を含有
- AD脳から単離したタウは3倍のリン酸を含有(vs. 健常脳から単離したタウ)
ミクログリアおよびアストロサイトの役割
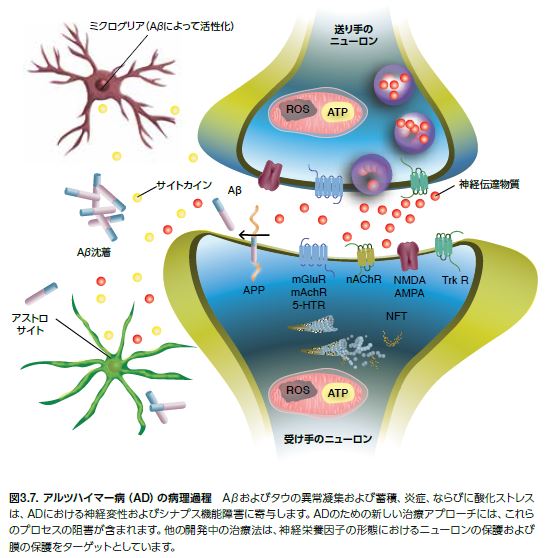
アストロサイト(BBBの血管を覆う) およびミクログリア(食細胞として機能する)を含むグリア細胞は、ADにおける神経変性プロセスに関与します。ニューロンおよびアストロサイトは連携して機能し、Aβ42オリゴマーを産生します。Aβダイマーおよびトリマーは、シナプス伝達の障害、酸化ストレスの誘導、タウの過剰リン酸化の促進に寄与します。また、ミトコンドリアに対し毒性を示し、ミクログリア細胞を活性化します。活性化ミクログリアは、神経細胞およびアストロサイトにおけるさらなるAβ42の産生を誘導するサイトカインおよび他の因子を分泌します。ミクログリア、神経炎症、およびADの関係はよく確立されています。その概略を図3.7に示します *30 。また、Aβ42オリゴマーは、ミエリンを含む、コレステロールの豊富な膜を損傷するというエビデンスがあります。Aβオリゴマーは、いくつかの異なるメカニズムによって除去されます。ネプリライシン(別名:膜メタロ-エンドペプチダーゼ(MME)およびインスリン分解酵素の2種類のプロテアーゼは、Aβを分解し、定常状態のAβレベルを低下させます。ネプリライシンレベルの低下は脳におけるAβ蓄積を導き、インスリン分解酵素の欠失はAβ分解を50%低下させ、いずれかの酵素の過剰発現はプラーク形成を減少させます。Aβオリゴマーは、アストロサイトおよびミクログリアによっても取り込まれ、受動的な流れを介して脳脊髄液に移行し、リポタンパク質受容体関連タンパク質は循環に取り込まれます *22,31。
 アルツハイマー病の正確な原因は不明ですが、アミロイドβプラークの形成がAD発症に関わっているという膨大なエビデンスが存在しています。
アルツハイマー病の正確な原因は不明ですが、アミロイドβプラークの形成がAD発症に関わっているという膨大なエビデンスが存在しています。
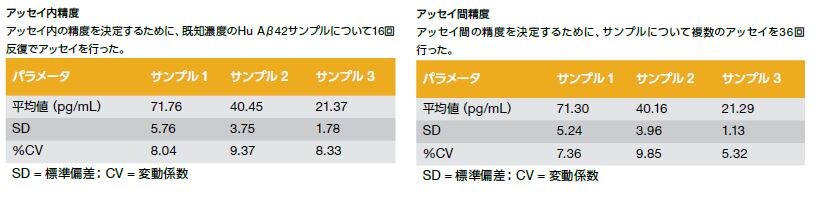
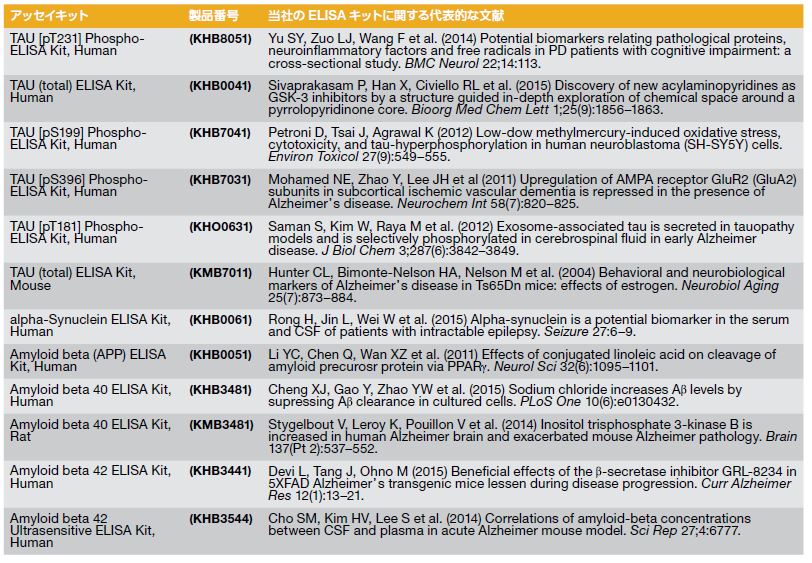
ADなどの神経変性状態について研究する研究者を支援するために、当社ではサンプル(例:細胞培養上清、組織ホモジネート、脳脊髄液(CSF)等)に含まれるヒトAβ42を定量するための、96ウェルプレートおよびマイクロプレートリーダーを用いる、研究使用限定の酵素結合免疫吸着測定法(ELISA)キットを提供しています。本キットは、ヒトAβ42の天然型および合成型を認識し、β40/Aβ43を検出せずに、Aβ42を選択的に検出することが可能です。(製品番号 KHB3544)
Amyloid beta 42 Ultrasensitive ELISA Kit, Human
性能特徴
- 感度: <1 pg/mL
- 標準曲線範囲:1.56-100 pg/mL
- サンプルタイプ:細胞培養上清、組織ホモジネート、CSF
- 種反応性:ヒト
- サンプル量:50 μL(希釈済み)
バリデーション
ELISAの完全なポートフォリオについてはこちらをご覧ください。
神経原線維変化の形成
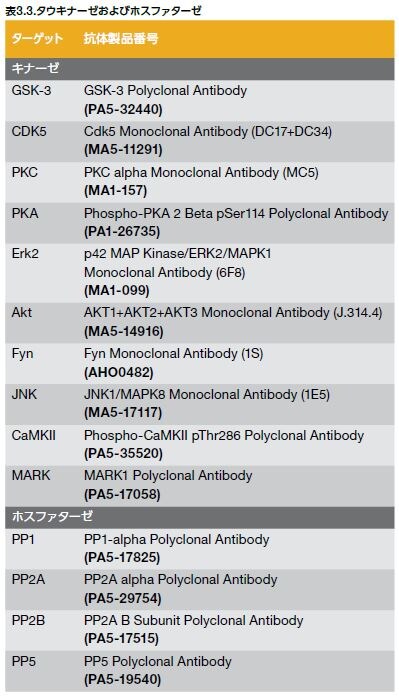
過剰にリン酸化されたタウは微小管から解離し、正常なタウのミスフォールディングを引き起こし、正常なタウ、MAP1およびMAP2を捕捉します。その結果、微小管の不安定化および細胞内におけるタウタンパク質のオリゴマー形成が生じます。神経原線維変化はタウのオリゴマー化の結果として起こり、軸索輸送障害および神経細胞のアポトーシスを誘導します(図3.8) *22,28 。GSK-3 およびCDK5はタウをリン酸化する主要なキナーゼです。タウは、表3.3および図3.8に示すような多くの他のキナーゼおよびホスファターゼによって、それぞれリン酸化および脱リン酸化されます *28 。
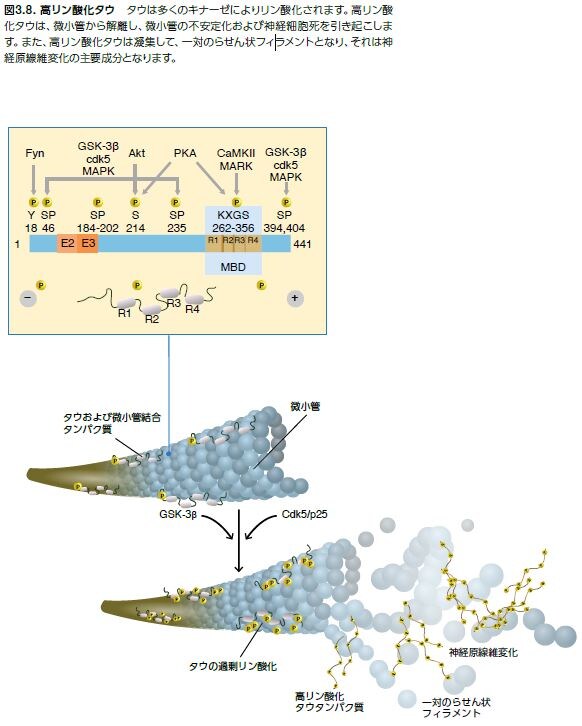
神経原線維変化の成分
神経原線維変化はほぼ全体的に高リン酸化タウから構成されていると一般的に誤認識されていますが、その構造はビメンチン、アクチン、ユビキチン、MAP2、アミロイドβ、およびタウなどの神経タンパク質に対する抗体で標識可能です *29。 神経原線維変化と共存することで知られているタンパク質を表3.4にリストアップします。

当社では、定量ソリューションとして、高品質の一次および二次抗体の幅広いポートフォリオに加えて、ELISA用試薬およびアクセサリ、単一ターゲット解析用の抗体ペア、ならびに神経生物学および他の研究分野用の簡便ですぐに使える完全なELISAキットを提供しています。神経生物学用ELISAに加えて、当社には下記の代表的ELISA製品カテゴリーがあります。
- リン酸ELISA
- サイトカインおよびケモカインELISA
神経生物学用ELISA
神経生物学用ELISAキットは、神経疾患の研究や神経科学関連の研究を支援するためにデザインされた高品質ELISAキットです。これらの“neuroELISA”キットは、脳脊髄液(CSF)、細胞培養上清、初代ニューロンライセート、および脳組織サンプルなどのサンプルタイプにおけるAβ、タウ、およびα-シヌクレインタンパク質の正確、高感度、かつ迅速な定量を可能とするようにデザインされています。
当社の神経生物学用ELISAキットの利点
- 数百件もの神経科学分野の査読論文に引用
- ELISA市場で25年の実績
- 厳格なバリデーション̶感度、ダイナミックレンジ、精度、特異性、回収率、およびロット間の一貫性などの業界基準の規格に適合し、信頼性のある結果を実現
- 簡便̶あらかじめコーティングされたプレート、捕捉抗体および検出抗体、スタンダード、バッファー、ならびにアクセサリー試薬を含む、ready-to-useのキット
 ELISAの完全なポートフォリオについてはこちらをご覧ください。
ELISAの完全なポートフォリオについてはこちらをご覧ください。
バイオマーカー
ADの組織学的および解剖学的特徴に関する基礎的な科学情報は、疾患検出や疾患進行のモニタリングのための臨床的方法を開発するために使用されてきました。現在では、数多くのイメージング法およびタンパク質バイオマーカーを検出するための技術がADの新しい治療法を開発するための臨床試験ならびに被験者の選択に日常的に使用されています。ADの臨床および実験室でのスクリーニングに使用される様々なバイオマーカーの概要を表3.5に示します *25。
バイオマーカーは、客観的に測定および評価できる指標あるいは実体であり、正常な生物学的過程、発病過程、または治療に対する反応を示します。バイオマーカーが疾患に特異的であるためには、その存在、濃度、および/または活性が疾患と関連付けられている必要があります *25,32 。結果的に、バイオマーカーは、疾患検出だけでなく、疾患進行のモニタリング、疾患発症の検出、治療に対する反応のモニタリング、さらに疾患の誤診を回避するためにも用いられます *25。
理想的なバイオマーカーの特徴を以下に示します。
- 高選択的(ほぼ全ての疾患サンプルで陽性を示す)
- 高特異的 (大部分の非疾患サンプルで陰性を示す)
- 疾患経過を正確に予測
- 治療に対する反応の程度を反映
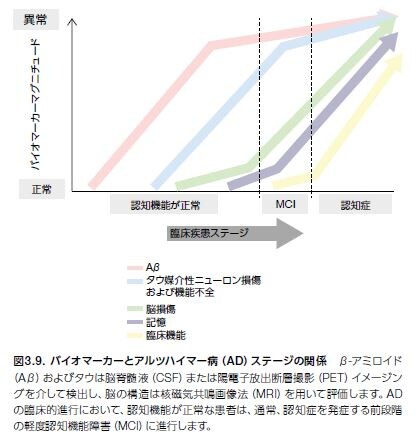
ADと診断された患者の圧倒的多数は、既に認知機能が損なわれています。現在のAD研究では、バイオマーカーを使用して、症状の発症前に疾患を検出するための研究が行われています。これは、疾患の初期段階に迅速かつ効果的な治療を行い、初期に疾患進行を抑制または遅延させる新しい治療法を開発することを最終目標とするために必要不可欠です *25。 生化学マーカーおよび構造マーカーならびに臨床症状の一時的な発現および蓄積を図3.9に示します。
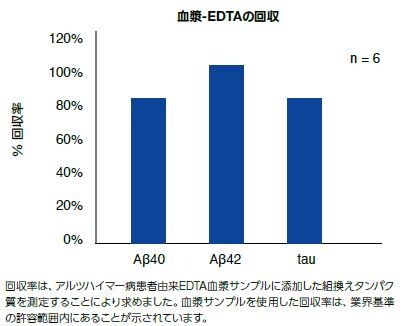
 Luminexマルチプレックス技術を用いたデータ出力の最大化アルツハイマー病研究の成功を促進するために、当社の新しいキットは、正確で一貫した、ヒト脳脊髄液中のAβ40、Aβ42および総タウの定量結果を提供するよう独自にデザインされています。組織ホモジネート、血漿、または組織培養上清などのサンプルタイプにも使用可能とされます。(製品番号 LHN0001M)
Luminexマルチプレックス技術を用いたデータ出力の最大化アルツハイマー病研究の成功を促進するために、当社の新しいキットは、正確で一貫した、ヒト脳脊髄液中のAβ40、Aβ42および総タウの定量結果を提供するよう独自にデザインされています。組織ホモジネート、血漿、または組織培養上清などのサンプルタイプにも使用可能とされます。(製品番号 LHN0001M)
Luminex™ PlatformのためのInvitrogen™ Amyloid beta (Aβ)/TauNeurodegenerative Human Magnetic3-plex Panelの導入
当社のキットの利点
- 3種類のターゲットの同時解析に必要なサンプルはわずか15 μL/well
- 感度、特異性、およびロット間の一貫性などの基準について厳格な試験を行い、再現性および正確な結果を実現
- マルチプレックス解析の効率、スピード、および広いダイナミックレンジと、ELISAと同等の結果を同時に実現
当社のキットは、Luminex™ 100™、200™、FLEXMAP 3D、またはMAGPIX装置において使用するように開発されています。
 完全なLuminexポートフォリオに関する情報についてはこちらをご覧ください。
完全なLuminexポートフォリオに関する情報についてはこちらをご覧ください。
当記事は、神経変性疾患研究ガイドブックからの抜粋です。
ガイドブックは下記から無料でダウンロード頂けます。
アルツハイマー病における神経炎症
神経炎症
近年まで、神経炎症はAD進行の後期にAβが蓄積するまで発生しないと考えられていました。近年、遺伝学およびバイオインフォマティクス研究により、免疫系の活性化はAD発症前の軽度認知機能障害の段階で生じ得ることが明らかにされました。免疫系タンパク質をコードする遺伝子に変異がある患者では、組織および体液における免疫メディエーターレベルの増加とADの初期症状との間に関係がみられることが観察されています。さらに、乾癬、肥満症などの症状でみられる高レベルの炎症を有する患者においては、ADを発症するリスクが増加します。累積証拠では、神経炎症がAD発症の原因となる影響を与えていることが示されています *33,34。
ミクログリア細胞
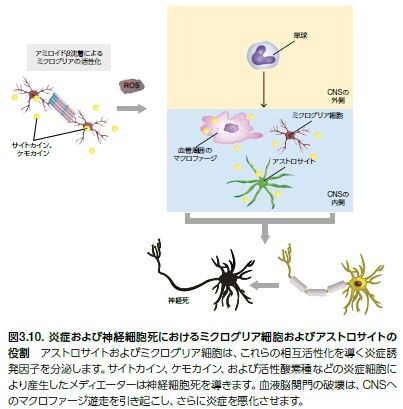
ミクログリア細胞̶自然免疫系の食細胞̶は、ADにおける神経炎症反応を導く主要な誘因です。この特殊化した細胞は、免疫監視の役割を担い、病原体や細胞デブリを除去し、記憶形成に関与する栄養因子を分泌します *33,34。 AD発病中の損傷、外傷、あるいはAβ沈着の蓄積はミクログリアの活性化を導く可能性があります。アストロサイトおよびミクログリア細胞は、これらの相互活性化を導く炎症誘発因子を分泌します。サイトカイン、ケモカイン、および活性酸素種などの炎症細胞により産生したメディエーターは神経細胞死を導きます(図3.10および表3.6)*34,35。
加齢およびADにおける血管炎症の証拠が数多くあるように、炎症は脳内だけでなく、BBBのレベルでも生じます。BBBの破壊は、血管腔からCNSに単球を移行させ、炎症環境をさらに悪化させます。また、AD患者の血管には、同年齢のコントロール患者と比較して、高レベルのIL-1β、IL-6、IL-8、TNF-α、TGF-β、およびMCP-1が含まれることが示されました *36 。

免疫療法
ミクログリア細胞は、サイトカインを分泌するだけでなく、抗原を貪食およびプロセシングして、T細胞に提示するための小さい断片とします。抗原提示はT細胞活性化の重要な要素で、細胞系譜に依存し、活性化T細胞は細胞を死滅させるか、免疫応答を改変するためのさらなるサイトカインを産生します。
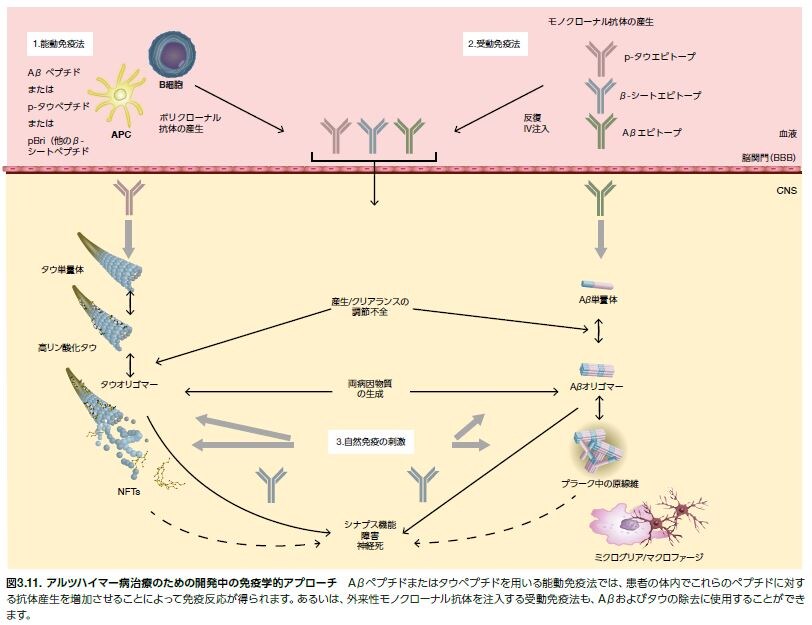
ADでは、Aβおよびタウが蓄積し、ミクログリア、B細胞、樹状細胞およびマクロファージなどの抗原提示細胞によってプロセシングされます *37。 Aβおよびタウを除去するための免疫系を利用する戦略が現在進行しています。研究者たちは、能動免疫療法あるいは受動免疫療法によってAβおよびタウのレベルを低下させ、それによりADの病状の進行を遅延または停止できることを期待しています(図3.11)。
Aβまたはタウをターゲットとするワクチンやヒト化モノクローナル抗体を用いる前臨床試験では、ADの病状の軽減と神経毒性の軽減に成功しています。しかしながら、臨床試験̶ADに対する能動免疫療法および受動免疫療法の両方のアプローチ̶では、有害反応(髄膜脳炎)、アミロイドイメージングの異常、および有効性エンドポイントに対する悪影響などの問題に遭遇しています *38,39 。現在は、ADの症発現前の投与やADに対する新しい免疫療法アプローチの開発における能動免疫法および受動免疫法の安全性および有効性の改善に焦点を当てて研究を進めています *38 。
実験的治療薬のためのターゲット
神経炎症と疾患の関係の理解に対する関心は高まってきています。神経免疫系の食細胞であるミクログリア細胞は、アルツハイマー病や他の神経変性疾患における脳の炎症反応に関わる鍵となる要素であると考えられます。活性酸素種に加えて、アストロサイトおよびミクログリア細胞は、神経細胞死を導く可能性のある炎症誘発性サイトカインおよび他の因子を分泌します。
当社では、神経炎症について研究するための抗体アレイを幅広く取り揃えています。当社の抗体は、免疫蛍光、免疫組織化学、ELISA、ウェスタンブロッティング、フローサイトメトリーおよび他のアプリケーションを含む幅広いアプリケーションでの使用についてバリデーション済みです。詳しい情報についてはこちらをご覧ください。
Invitrogen™ Attune™ NxT Flow Cytometer:パワフルでフレキシブル
- モジュール式デザイン̶フィールドアップグレード対応の多様な機器構成をラインナップ対応
- 所要時間を短縮̶データ品質を維持したまま10倍に高速化
- 簡潔なサンプル調製̶洗浄不要・溶血不要オプションによる目詰まり防止型の流体工学を採用
- ユーザー独自の用途に対応̶あらゆる細胞型とサンプルを扱う複雑なプロトコルに対応
表現型解析におすすめのサイトメーターに関する詳しい情報についてはこちらをご覧ください。
アルツハイマー病の新しい治療法
アルツハイマー病の進行を回復または遅延させるための疾患修飾治療は現在存在しません。既存の治療は、認知機能低下を遅延させることにより一時的に症状を改善するもので、神経終末の神経伝達を改善することに焦点が当てられています。1つの薬剤クラスでは、アセチルコリン分解酵素であるアセチルコリンエステラーゼ(AChE)の阻害を介して神経終末のアセチルコリン(ACh)レベルを増加させます(シナプスからAChを放出させるための主要なメカニズム)(図3.11)。AD治療薬としてFDAに承認されているAChE阻害薬は、リバスチグミン、ドネペジル、およびガランタミンの3つです。さらに近年、NMDA受容体拮抗薬であるメマンチンがAD治療薬として承認されました(図3.11)。メマンチンは、グルタミン酸興奮毒性を抑制すると考えられます *8 。
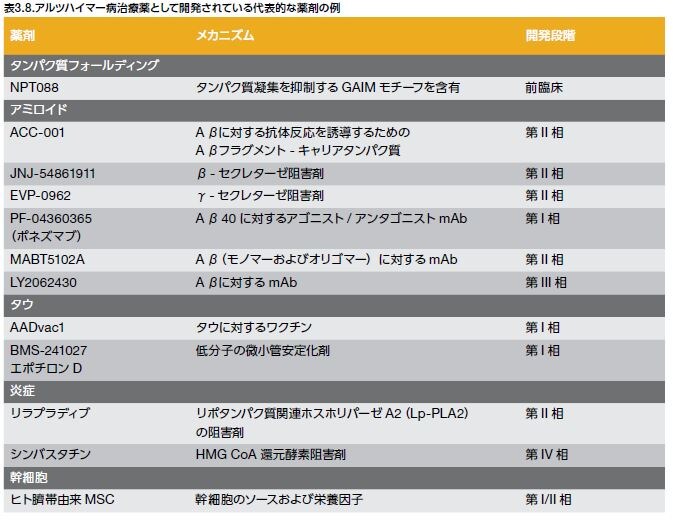
ADの認知症状の治療に対して承認されている治療薬の作用機序は2種類だけですが、数多くの有望な実験的アプローチが行われています。ADに対する実験的アプローチの例としては、以下の戦略―アミロイドβおよびタウをターゲットとする薬剤の開発、CNSにおけるROS、アポトーシス、および炎症を阻害する薬剤の開発、ならびに 神経保護および再生を促進する細胞ベースの治療の実現可能性の評価―が挙げられます。現在臨床試験中の代表的な薬剤を表3.8に示します *8,27,31,40-42。
アルツハイマー病に対する新しい幹細胞ベースの治療
ADに対する新しい薬剤および実験的な免疫療法戦略に加えて、幹細胞研究分野はこの壊滅的な疾患に対し切望されている治療法を提供するものとして非常に有望視されています。現在では、技術の進歩により、高純度かつ高濃度の神経幹細胞およびグリア幹細胞の生産が可能です。神経幹細胞(NSC)は、胎児脳組織、新生児および成人の死後脳組織、胚性幹細胞(ESC)、あるいは人工多能性幹細胞(iPSC)などの数多くのソースから獲得することができます。
げっ歯類の試験では、NSC移植後にコリン作動性ニューロンの増加および記憶の改善が示されましたが、この改善は、移植されたNSCが既存の組織に組込まれた結果か、NSCの栄養因子分泌によるものか明らかではありません。ADラットモデルを用いた一連の試験では、ヒト神経成長因子遺伝子を発現しているラットNSCは生存し、大脳皮質に組込まれ、認知行動が増加しましたが、ヒトNGF遺伝子を含まないNSCは生存せず、大脳皮質に組込まれず、認知機能が改善しませんでした *2。
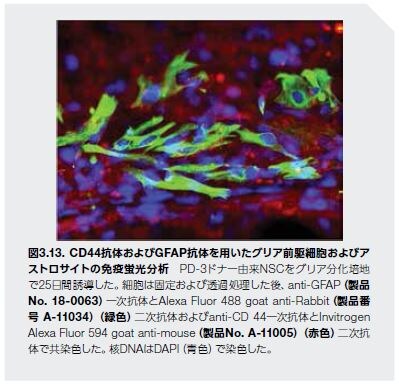
前臨床試験用の幹細胞を作製および分離する能力は、新しい臨床治療として幹細胞を研究するための道を切り開きます。ADに対する最初の幹細胞臨床試験は最近開始され(図3.13)、注入された幹細胞のMRIによる追跡に成功した例もあります。AD患者由来の幹細胞は、有望なAD治療法として開発されているだけでなく、新しい薬理療法をスクリーニングするための実験モデルとしても使用されています *43。
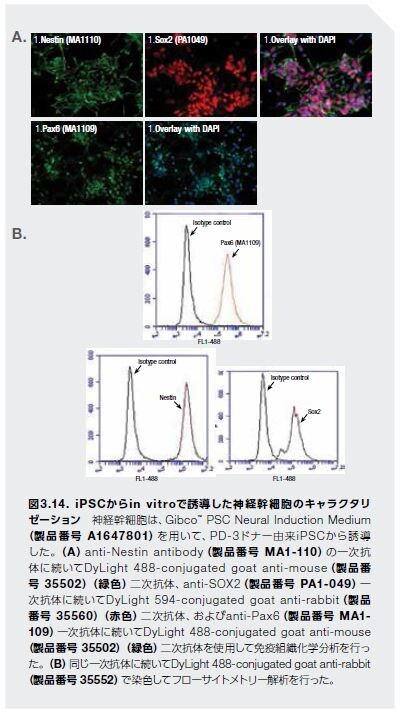
神経変性疾患のための抗体ベースのツール
生物医学研究のためのイムノアッセイ用の高品神経生物学および他の研究領域の分野における新しい発見に貢献します。当社では、ELISAキット、Luminex装置プラットフォーム用のマルチプレックスアッセイ、および抗体ペアキットの幅広いセレクションを、簡便ですぐに使えるフォーマットで提供しています。当社の提供するタンパク質定量のためのイムノアッセイの質に関する詳しい情報、およびタンパク質定量ガイドの無料ダウンロードについてはこちらのサイトをご利用ください。
当社のELISAキットは3,000件以上の論文で参照されており、マルチプレックスアッセイは1,000件を超える論文に引用されています。当社では、高感度で特異的な、細胞内または細胞外タンパク質の検出のために、広範囲な免疫ベースの製品を、簡便ですぐに使えるフォーマットで提供しています。当社では、単一分析物の解析用の抗体ペアキットおよびELISAキット、さらに複数ターゲット同時解析用のマルチプレックスアッセイを開発 しました。当社のキットは、感度、特異性、精度、およびロット間の一貫性などの基準に関して厳格なバリデーションプロセスを通過しており、信頼性のある正確な結果を提供します。神経変性疾患研究および他の領域の生物医学研究には、Invitrogen™およびThermo Scientific™のイムノアッセイをご利用ください。
新しいガイドに必要とする製品が記載されていなくても心配ご無用です。̶当社は、幅広いカスタム製品開発サービスも提供しています。こちらをご覧ください。
【無料ダウンロード】神経変性疾患研究ガイドブック
当記事は、神経変性疾患研究ガイドブックからの抜粋です。以下の内容を含むPDFは下記から無料でダウンロード頂けます。
~主な内容~
- イントロダクション
- 神経変性疾患における共通のテーマ
- アルツハイマー病への取り組み
- 他の主要な神経変性疾患
- 抗体:神経変性疾患研究のための強力なツール
参考文献:
1. Stages of Alzheimer’s. alz.org. http://www.alz.org/alzheimers_disease_stages_of_alzheimers.asp. Accessed August 2015.
2. Tong LM, Fong H, Huang Y (2015) Stem cell therapy for Alzheimer’s disease and related disorders: current status and future perspectives. Exp Mol Med 13;47:e151.
3. Bird TD. Alzheimer Disease Overview. 1998 [Updated 2014 Apr 3]. In: GeneReviews [Internet]. Pagon RA, Adam MP, Ardinger HH, et al., editors. Seattle (WA): University of Washington, Seattle; 1993-2015. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1161/. Accessed August 2015.
4. 2015 Alzheimer’s Disease Facts and Figures. Alzheimer’s association. https://www.alz.org/facts/downloads/facts_figures_2015.pdf. Accessed August 2015.
5. Sheng M, Sabatini BL, Südhof TC (2012) Synapses and Alzheimer’s disease. Cold Spring Harb Perspect Biol 1;4(5).
6. Ray M, Zhang W (2010) Analysis of Alzheimer’s disease severity across brain regions by topological analysis of gene co-expression networks. BMC Syst Biol 4:136.
7. Moreira PI, Carvalho C, Zhu X et al. (2010) Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim Biophys Acta 1802(1):2–10.
8. Guzior N, Wieckowska A, Panek D et al. (2015) Recent development of multifunctional agents as potential drug candidates for the treatment of Alzheimer’s disease. Curr Med Chem 22(3):373–404.
9. Collingwood J Brain Fluid Changes Predict Alzheimer’s 10 Years in Advance. Psych Central. http://psychcentral.com/lib/brain-fluid-changes-predictalzheimers-10-years-in-advance/. Accessed August 2015.
10. Gendelman HE, Anantharam V, Bronich T et al. (2015) Nanoneuromedicines for degenerative, inflammatory, and infectious nervous system diseases. Nanomedicine 11(3):751–767.
11. 2015 Alzheimer’s Statistics. Alzheimers.net. http://www.alzheimers.net/resources/alzheimers-statistics/. Accessed September 2015.
12. 2015 Alzheimer’s Statistics. Alzheimers.net. http://www.alz.org/dementia/typesof-dementia.asp. Accessed August 2015.
13. Alzheimer’s Disease Fact Sheet. National Institute on Aging. https://www.nia.nih.gov/alzheimers/publication/alzheimers-disease-fact-sheet. Accessed August 2015.
14. Lott IT, Head E (2005) Alzheimer disease and Down syndrome: factors in pathogenesis. Neurobiol Aging. 2005 Mar;26(3):383–389.
15. Barber RC (2012) The genetics of Alzheimer’s disease. Scientifica (Cairo) 2012:246210.
16. Risk Factors. Alz.org. http://www.alz.org/alzheimers_disease_causes_risk_factors.asp. Accessed August 2015.
17. Serrano-Pozo A, Frosch MP, Masliah E (2011) Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 1(1):a006189.
18. Apostolova LG, Green AE, Babakchanian S et al. (2012) Hippocampal atrophy and ventricular enlargement in normal aging, mild cognitive impairment (MCI), and Alzheimer Disease. Alzheimer Dis Assoc Disord 26(1):17–27.
19. Rosso AB, Inestrosa NC (2013) WNT signaling in neuronal maturation and synaptogenesis. Front Cell Neurosci 7:103.
20. Pescosolido N, Pascarella A, Buomprisco G et al. (2014) Critical review on the relationship between glaucoma and Alzheimer’s disease. Adv Ophthalmol Vis Syst 1(4):00024.
21. Zhang X, Li Y, Xu H et al. (2014) The -secretase complex: from structure to function. Front Cell Neurosci 8:427.
22. Querfurth HW, LaFerla FM (2010) Alzheimer’s disease. N Engl J Med 362(4):329–344.
23. Sakono M, Zako T (2010) Amyloid oligomers: formation and toxicity of Abeta oligomers. FEBS J 277(6):1348–1358.
24. Goure WF, Krafft GA, Jerecic J (2014) Targeting the proper amyloid-beta neuronal toxins: a path forward for Alzheimer’s disease immunotherapeutics. Alzheimers Res Ther 6(4):42.
25. Anoop A, Singh PK, Jacob RS et al. (2010) CSF biomarkers for Alzheimer’s disease diagnosis. Int J Alzheimers Dis 23;2010:606802.
26. Mandelkow E, Mandelkow EM (1995) Microtubules and microtubule-associated proteins. Curr Opin Cell Biol 7(1):72–81.
27. Boutajangout A, Wisniewsik T (2014) Tau-based therapeutic approaches for Alzheimer’s disease – a mini-review. Gerontology. 2014;60(5):381–385.
28. Wang JZ, Xia YY, Grundke-Iqbal I et al. (2013) Abnormal hyperphosphorylation of tau: sites, regulation, and molecular mechanism of neurofibrillary degeneration. J Alzheimers Dis 33 Suppl 1:S123–S139.
29. Wischik CM, Harrington CR, Storey JM (2014) Tau-aggregation inhibitor therapy for Alzheimer’s disease. Biochem Pharmacol 88(4):529–539.
30. Aso E, Ferrer I (2013) Potential Therapeutic Strategies to Prevent Progression of Alzheimer to Disease States. Chapter 11 in: Understanding Alzheimer’s Disease. Intech http://cdn.intechopen.com/pdfs-wm/43125.pdf. Accessed August 2015.
31. Kumar A, Singh A, Ekavi AS (2015) A review on Alzheimer’s disease pathophysiology and its management: an update. Pharmacol Rep 67(2):195–203.
32. Puntmann VO (2009) How-to guide on biomarkers: biomarker definitions, validation and applications with examples from cardiovascular disease. Postgrad Med J 85(1008):538–545.
33. Heppner FL, Ransohoff RM, Becher B. (2015) Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci 16(6):358–372.
34. Heneka MT, Carson MJ, El Khoury J et al. (2015) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14(4):388–405.
35. Morales I, Guzmán-Martínez L, Cerda-Troncoso C et al. (2014) Neuroinflammation in the pathogenesis of Alzheimer’s disease. A rational framework for the search of novel therapeutic approaches. Front Cell Neurosci 22;8:112.
36. Marques F, Sousa JC, Sousa N et al. (2013) Blood-brain-barriers in aging and in Alzheimer’s disease. Mol Neurodegener 22;8:38.
37. Li Y, Tan MS, Jiang T et al. (2014) Microglia in Alzheimer’s disease. Biomed Res Int 2014:437483.
38. Yang C, Xiao S (2015) New developments of clinical trial in immunotherapy for Alzheimer’s disease. Curr Pharm Biotech16(6):484–491. http://www.ingentaconnect.com/content/ben/cpb/2015/00000016/00000006/art00002?token=00431021437a63736a6f35475f4c7a763c24452a5b6f644a467b4d616d3f4e4b342. Accessed August 2015.
39. Wisniewski T, Go i F (2015) Immunotherapeutic approaches for Alzheimer’s disease. Neuron 18;85(6):1162–1176.
40. Han SH, Mook-Jung I (2014) Diverse molecular targets for therapeutic strategies in Alzheimer’s disease. J Korean Med Sci 29:893–902.
41. Kang JH, Ryoo NY, Shin DW et al. (2014) Role of cerebrospinal fluid biomarkers in clinical trials for Alzheimer’s disease modifying therapies. Korean J Physiol Pharmacol 18(6):447–456.
42. Jia Q, Deng Y, Qing H et al. (2014) Potential therapeutic strategies for Alzhiemer’s disease targeting or beyond -amyloid: insights from clinical trials. BioMed Res Int 837157
43. ALZFORUM/Networking for a Cure. Ready or Not: Stem Cell Therapies Poised to Enter Trials for Alzheimer’s. http://www.alzforum.org/news/conferencecoverage/ready-or-not-stem-cell-therapies-poised-enter-trials-alzheimers. Accessed August 2015.
研究用にのみ使用できます。診断目的およびその手続き上での使用はできません。
記事へのご意見・ご感想お待ちしています

予測ゲノミクスでがんを理解する
この記事は、科学者向けにがん研究の新しい...
Read More
神経科学におけるフラグメント解析:複数の疑問を解決する柔軟なツール
脳は驚くべき器官です。さらに驚くべきこと...
Read More
リキッドバイオプシーを活用した肺がんと悪性脳腫瘍に対する研究事例のご紹介
リキッドバイオプシーは血液や尿などの体液...
Read More
細胞培養関連の学習リソースまとめ~トレーニング・セミナー・ハンドブックなど~
「細胞培養」はライフサイエンス研究におけ...
Read More