1つの神経変性症状を他の明確に異なる症状と識別し分類するための境界線はよく認識されています。認知機能障害および運動機能障害の患者は、高頻度で1つ以上の明確な神経変性疾患の神経学的病状が疑われ、また複数の病態マーカーの存在は神経学的障害の増加と関係します。しかしながら、ケースによっては、病態マーカーで検出された症例に明白な障害の兆候が見られないこともあります。このような状況において、決定的な疾患分類を確立することは、神経変性疾患の診断、治療、および研究における課題です。
アルツハイマー病、レビー小体型認知症、パーキンソン病、および他の症状などの様々な症候群の間には、高頻度にオーバーラップが存在するにも関わらず、この疾患グループを定義するために伝統的に使用されている明確な病態マーカーを評価することは、研究者が神経毒性および神経変性に寄与する発症機序の解明を始めることを可能とするフレームワークを提供することになります。
※本記事の末尾において、神経変性疾患について体系的に学べる無料コンテンツを紹介しています。
パーキンソン病
パーキンソン病の特徴
パーキンソン病(PD)は、運動機能不全を特徴とする進行性の神経変性疾患です *1 。PDは、パーキンソニズムの最も一般的な形態で、筋振戦および筋固縮、動作緩慢、姿勢の不安定、ならびに歩行障害によって特徴付けられる全ての臨床障害を包含します。PDを発症した患者の5人に1人は認知症も発症します。孤発性PDの症状は一般的に60歳前後で発症しますが、若年発症者はこの疾患の遺伝型を持っていることが考えられます。PDは、非対称性四肢硬直やレボドパ(L-3,4-ジヒドロキシフェニルアラニン;L-DOPA)による治療に対する反応などの臨床所見に基づいて診断されます。2,3 L-DOPAまたはドーパミンアゴニストによる治療により 運動症状は改善されますが、疾患進行を変化させることはできません(図4.1)。疾患の経過において、大部分の患者は、乾癬、抑うつ症状、認知症、自律神経障害、睡眠障害、および疼痛などの非運動性の症状を経験します *1 。
PDの遺伝子変異体
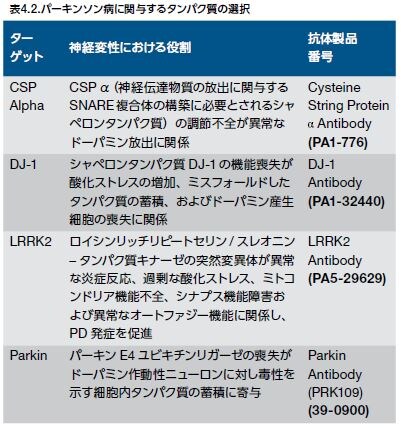
大部分のPD患者は孤発性疾患ですが、わずか10%の患者には家族歴が存在します。これにも関わらず、約20年間の研究によって、優性または劣性の遺伝形式を持つ数種のPDの単一遺伝子形態が発見されました。PDは、PDとの関連性を示すPARKと呼ばれる特定の遺伝子座に基づいて分類することができます。単一遺伝子性のPDでは、6種類の遺伝子が確認されています。このうち、SNCA(PARK1)およびLRRK2(PARK8)は、いずれも常染色体優性の遺伝形式を示し、parkin(PARK2)、PINK1(PARK6)、DJ-1(PARK7)、およびATP13A2(PARK9)は、劣性の遺伝形式を示します(表4.1) *4-6 。 他の遺伝子は、PD発症の単独の要因とはなりませんが、PDを発症させるリスクを増加させる可能性があります。例えば、GBA1遺伝子はリソソームグルコセレブロシダーゼをコードしますが、GBA遺伝子における変異は孤発性PDの発症リスクの増加に関連するタンパク質機能を低下させます *4,7,8 。
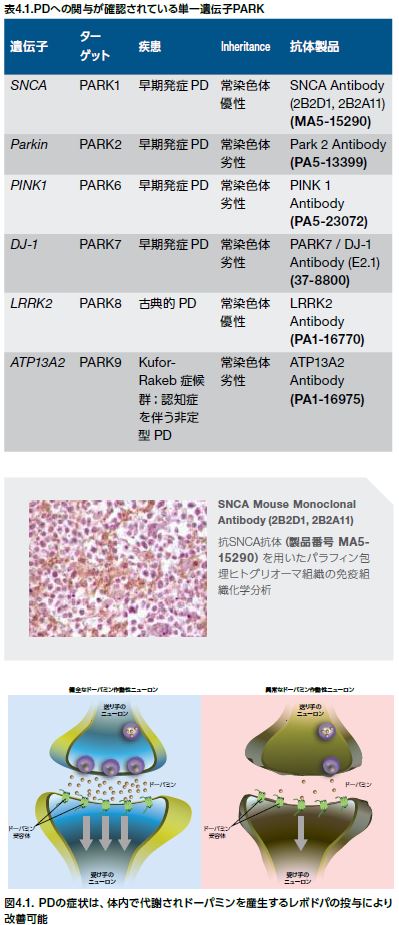
PDの顕著な特徴
数多くの様々な疾患がPDと共通する特徴を持ちますが、孤発性PDに関連する重要な特徴は黒質におけるドーパミン作動性ニューロンの喪失とレビー小体の存在、主にα-シヌクレインからなる細胞質内凝集体です *2 。PD患者由来の死後脳の研究において、α-シヌクレインの免疫反応性が細胞体および不溶性線維としての神経突起を通して確認され、それぞれレビー小体およびレビー神経突起として可視化されました。α-シヌクレインの凝集は、PD患者の広範囲の脳領域で検出され、アルツハイマー病およびレビー小体型認知症などの他のシヌクレイン病にも存在します*9,10。
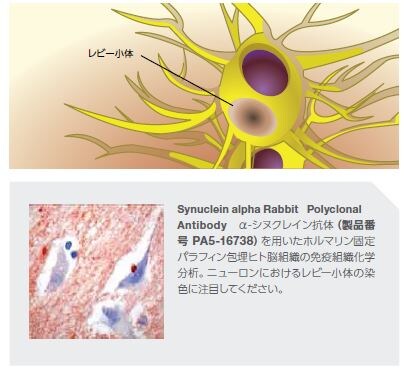
α-シヌクレイン
α-シヌクレインは、ホスホリパーゼD2を阻害する神経シナプス前タンパク質で、SNCA遺伝子(PARK1遺伝子座)にコードされています。α-シヌクレインに対する抗体は、α-シヌクレインがレビー小体の主要成分であることを決定するために使用されてきました。*9,11 野生型α-シヌクレインは線維凝集体を形成し、多くの様々なα-シヌクレイン変異体は、増強されたレビー小体形成能を示します *12 。α-シヌクレイン遺伝子の過剰なコピーや特定のミスセンス変異(A53TおよびA30P)は家族性PDの早期発症に関係します *12 。α-シヌクレインのタンパク質ミスフォールディングは、野生型タンパク質の過剰発現、野生型α-シヌクレインの発現に対する反応、ならびに酸化的ストレッサーおよび炎症に対する反応として生じる可能性があります。近年の研究では、チロシンキナーゼc-Ablのレベルおよび活性の増加がα-シヌクレイン蓄積の増加を導いていることが示唆されています。c-Abl阻害薬であるニロチニブ(FDA承認済みの慢性骨髄性白血病治療薬)は、PDマウスモデルにおいて神経保護作用およびドーパミンレベルの回復作用を示しました *13,14 。
病態生理学
野生型α-シヌクレインは、シナプス伝達の調節および神経終末からの神経伝達物質の放出に関与している可能性が示唆されています *15 。また、α-シヌクレインは神経保護の役割も担っていると考えられます。例えば、α-シヌクレインを発現させ、シャペロンタンパク質であるシステイン-ストリング-タンパク質-α(CSPα)を欠損させたトランスジェニックマウスでは、神経変性の早期発症および神経細胞死からレスキューされました。反対に、α-シヌクレインノックアウトモデルでは、CSPα媒介神経変性が悪化しました *16 。野生型α-シヌクレインは神経保護作用を有するとされるだけでなく、α-シヌクレインの凝集はドーパミン作動性ニューロンの変性に関係し、最終的に無数の運動神経の後遺症を引き起こすという数多くのエビデンスが存在します *17。
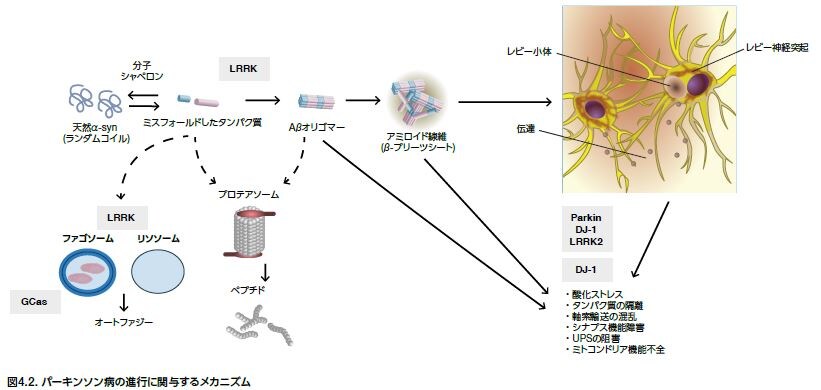
変異および環境因子は、オリゴマーを形成するミスフォールドしたα-シヌクレインタンパク質の産生を導きます。これらの異常タンパク質は蓄積し、正常なリソソームおよびプロテアソームの除去機構を回避します。α-シヌクレインの凝集は、酸化ストレスを誘導し、軸索輸送を障害し、シナプス機能障害を起こし、ミトコンドリア機能不全を促進することが示されています。疾患の進行は、部分的に、ミスフォールドしたタンパク質がニューロンからニューロンへと広がることによって生じると考えられます(図4.2) *10 。
既存の治療法
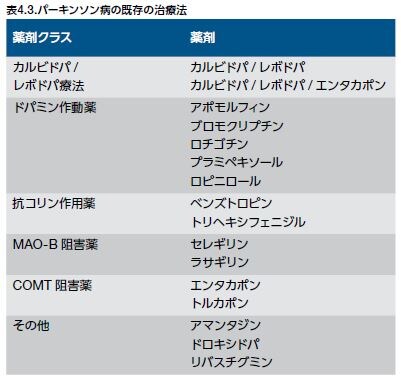
PDに関連する運動症状は、黒質におけるドーパミン作動性ニューロンの喪失が原因です。既存の治療アプローチの多くは、ドーパミン神経伝達の増強を目標としています(表.3) *18 。このような戦略は非常に効果的ですが(特に疾患発症時)、時間の経過とともに、これらの治療法は一連の運動性の副作用を伴います。また、PDの進行に伴い、非ドーパミン作動性の運動症状および非運動症状が顕著になってきます。このため、将来のPD治療法の探索は、神経保護および疾患修飾を戦略とします *19 。
実験的治療
パーキンソン病の新しい治療戦略としては、炎症、酸化、アポトーシス、およびタンパク質のミスフォールディング/凝集などの疾患プロセスの阻害が挙げられます。これらの治療アプローチについては、PDの疾患修飾療法として探索されてきているか、現在探索中ですが、幹細胞を使用した新しい技術を用いる治療法の一部では臨床試験が開始されています。また、リプログラミング因子の異所性発現を介して患者の成熟細胞由来の人工多能性幹細胞(iPSC)を作製する能力は、幹細胞研究に変革をもたらしています *20 。将来的には、患者由来iPSCを神経変性疾患モデルや治療薬の開発に使用するだけでなく、PD患者に対する細胞ベースの療法としての開発の可能性も秘めています *19,21-22 。
 当社の抗体ベースツールの幅広いポートフォリオについてはこちらをご覧ください。
当社の抗体ベースツールの幅広いポートフォリオについてはこちらをご覧ください。
当記事は、神経変性疾患研究ガイドブックからの抜粋です。
ガイドブックは下記から無料でダウンロード頂けます。
ハンチントン病
ハンチントン病の特徴
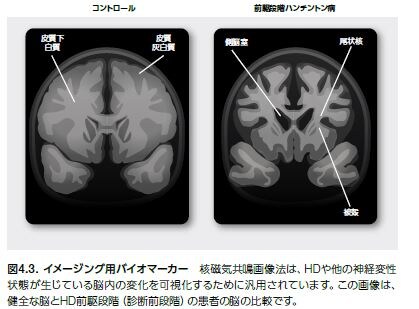
ハンチントン病(HD)は、進行性で、通常遺伝性の、典型的な三大症状を特徴とする神経変性疾患です(表4.4)*23 。症状の平均発症年齢は約35~44歳ですが、10%の症例では幼児期に発症します *24,25 。ハンチントン病の生存期間の中央値は一般的には発症年から15~18年です *27 。現在、HDの進行を遅延または停止させる治療は存在しません *28 。本疾患の自然経過には、明らかな疾患に先行して微妙な変化を来たす“前駆”段階も存在します。(図4.3) *25 。
研究の重要な新領域の1つは、前駆段階における微妙な変化を検出するバイオマーカーの同定です。早期の医療介入につながり、創薬努力を前進させる早期発見を可能とし、さらに疾患進行の追跡も可能とするバイオマーカーの開発は急務とされています。これらのバイオマーカーは、早期医療介入のための治療法の提案を可能とします *26 。
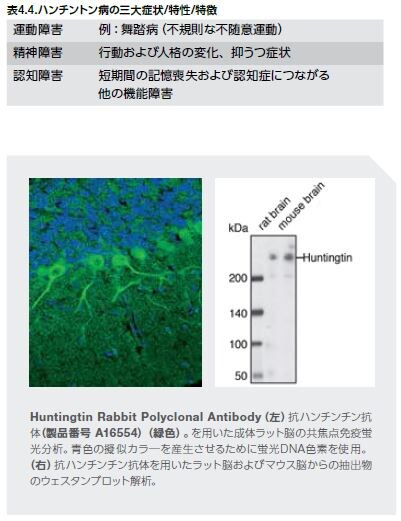
HDの原因
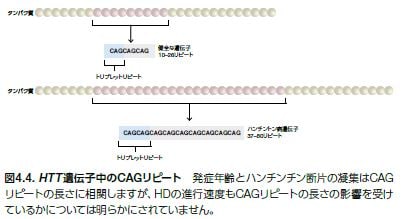
HDの遺伝型は常染色体優性遺伝形式を示すため、1つの異常なハンチンチン遺伝子(HTT)アレルが受け継がれた子孫にこの疾患が発現します。孤発性(非遺伝性)のHD症例は稀で、5~8%のHD症例にみられます *25 。HDは、HTT遺伝子領域をコードするN末端に存在するCAGリピートの異常伸長を原因とするCAGリピート病に分類されます。CAGリピートは、ポリQ伸長と呼ばれる連続するグルタミン(Q)残基をコードします。その長さは疾患の浸透率と病原性を決定します(図4.4)*23,25 。発病に至る伸長をしたCAGアレルは、神経細胞死を引き起こす神経毒性を持つ断片化された凝集体を形成するハンチンチンタンパク質を産生する機能獲得型変異として作用します *25 。
病態生理学
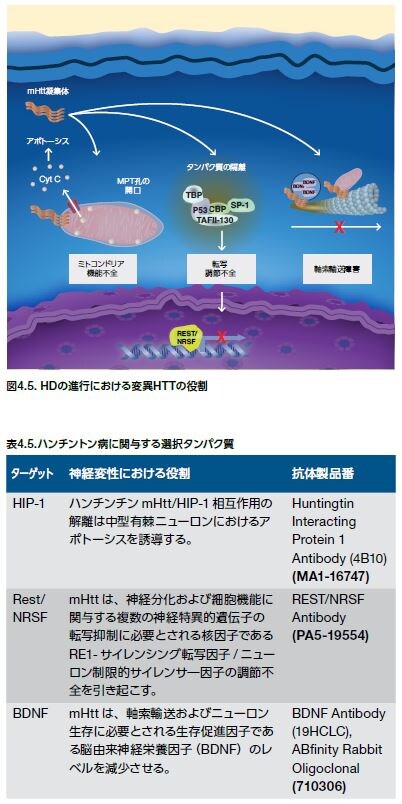
広範囲にわたる神経損傷および皮質変性は、認知障害および人格変化の主な原因です。大脳基底核̶特に、淡蒼球(GPeおよびGPiニューロン)ならびに線条体̶のニューロンの喪失は、舞踏病の主な原因です *29,30 。線条体におけるニューロンの喪失は、運動機能を制御する2つの皮質-線条体-視床回路(皮質→線条体→視床)を障害します。これらの回路(直接路および間接路)は、運動皮質に信号を送る視床で合流します。パーキンソン病とは対照的に、黒質のドーパミン作動性ニューロンは影響を受けないため、実際、HD患者の線条体におけるドーパミンレベルは正常値より高いです *31 。ドーパミンレベルの上昇は舞踏病にも寄与します。
細胞内で、ハンチンチンは、完全長タンパク質としても、タンパク質分解や選択的スプライシング由来のタンパク質断片としても存在します。これらの断片の形成̶特にハンチンチンの最初の100アミノ酸を含むN末端断片(例:HTTエキソン1モノマー)̶は、ハンチントン病の病態形成に必要とされます。変異ハンチンチン(mHtt)凝集体は、ニューロン遺伝子の転写、軸索輸送、およびミトコンドリア機能に悪影響を与えます(図4.5)。ポリQ拡張は、自己凝集を毒性形態に促進させるこれらの断片に代わるコンフォメーションをもたらすと考えられます *25 。ポリQ拡張はN末端断片と、神経細胞における小胞輸送の役割を担うハンチンチン相互作用タンパク質1 (HIP-1)のような結合パートナーやmHttによって調節不全される他のタンパク質との相互作用の仕方も変化させる可能性があります。(表4.5)*25。
ターゲットおよび有望な治療薬
既存の治療は、疾患を予防または遅延させるよりも、症状の重篤度を軽減させるために使用されています。シナプス小胞アミントランスポーター阻害剤かつドーパミン経路阻害剤であるテトラベナジンは、ハンチントン病に伴う舞踏運動に対する唯一のFDA承認薬です *25 。HDに対する新しい治療アプローチの開発の一環としてのバイオマーカーの同定は、HDの存在および重篤度の評価を可能とし、薬剤の活性および副作用に関する情報を提供し、無症状期または前駆期の期間に疾患を予測するマーカーを同定します。これらのバイオマーカーは治療のためのターゲットも同定します。既存の有望な新しいバイオマーカーは、HD患者および対照群の個体間の認知的、電気生理学的、解剖学的、生化学的、あるいは分子的な、臨床特性の違いを検出します。例えば、HD患者のCSFではIL-6、IL-8、およびTNF-αなどのサイトカインレベルの上昇がみられる可能性があるのに対し、HD患者の特定の白血球では脂肪酸アミド加水分解酵素や他の因子のレベルの減少がみられる可能性があります *25,32 。新しい治療アプローチは、毒性を持つHttタンパク質凝集体の定常状態レベルの減少や、mHtt凝集体によって障害される細胞プロセスの調節を目的として開発中です。基礎研究/前臨床段階のこれらの戦略の代表的な概要を表4.6に示します *31,33,34 。
幹細胞ベースの治療
新しい治療戦略に加えて、これらの最先端のアプローチでは、内在性幹細胞の特定のタイプ(例:間葉系幹細胞)を使用したり、ヒト人工多能性幹細胞やヒト胚性幹細胞株を活用します。最終目標は、1)正常な脳回路の再生を目的として移植するためのiPSCまたはhESC幹細胞の特定の神経細胞タイプまたはグリア細胞タイプへの分化、あるいは2)BDNFなどの神経栄養因子の効率的なソースとしての幹細胞の使用です *35 。
第1の目標のための重要要件は、被験者に移植するために、幹細胞を、明確に定義され、高度に純化された系列決定済み細胞または分化細胞の集団に分化させる能力です。この目標を達成するために、2つの補完的なアプローチが使用されています。トランスクリプトームアプローチによって、固有の遺伝子サインや遺伝子発現パターンに基づいて、幹細胞、前駆体、および分化した神経細胞またはグリア細胞の特性決定および分類を行います。プロテオームアプローチは、タンパク質発現サインを検出しますが、細胞表面マーカーおよび栄養/分化因子を同定するために使用できる点でトランスクリプトームアプローチよりも効率的です *35 。
 神経変性疾患研究を促進する抗体ベースのツールについてはこちらをご覧ください。
神経変性疾患研究を促進する抗体ベースのツールについてはこちらをご覧ください。
当記事は、神経変性疾患研究ガイドブックからの抜粋です。
ガイドブックは下記から無料でダウンロード頂けます。
筋委縮性側索硬化症
筋委縮性側索硬化症の特徴
筋委縮性側索硬化症(ALS)は、ルー・ゲーリック病とも呼ばれ、脳および脊髄の運動ニューロンが侵されます。ALSは、成人後に発症する最も一般的な運動ニューロン疾患で、3番目に一般的な神経変性疾患です *36 。ALSの症状には、反射の変化、筋力低下、筋肉の萎縮、攣縮、および痙攣があります。ALSは、一般的に約60歳で発症し、1つの筋肉サブセットにおいて徐々に症状が始まり、他の筋肉グループに派生していきます *37 。80%の症例において脊髄に発症し、非対称性で無痛の四肢の筋力低下が生じます *37 。また、20%の症例において咀嚼、嚥下、および発声を司る顔面の延髄筋に発症します。ALS患者は、症状の発症後、平均3年生存しますが、生存期間には顕著なばらつきもあります *41 。死亡原因は、通常、呼吸困難および呼吸の運動制御障害です *41。 ALSでは、運動系の障害に加えて、認知機能の変化が認められることがあります。前頭側頭葉の機能障害は、軽度の異常から重篤な前頭側頭葉認知症(FTD)の範囲で発現します。ALSとFTDの両方を発現した患者は、いずれかを単独で発現した患者に比べて生存率が低下します *44 。
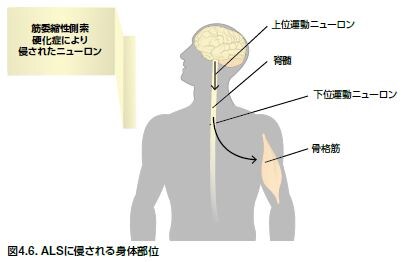
一般的原因
ALS症例のうち、約10%が家族性ALSであり、全部ではありませんが、大部分の家族性ALSは単一遺伝子変異の継承によって発症します *37 。遺伝的原因については、現在では約3分の2の家族性ALS症例と10%の孤発性ALSについて明らかにされています *36 。60%のALSの症例は、SOD1、TARDBP/TDP-43、FUS、およびC9orf72のわずか4種類の遺伝子の変異によって発症しています *36 。異なるALS変異が同じ細胞プロセスに影響しているのではという新しいテーマが提起されています。例えば、SQSTM1およびTBK1遺伝子はともにオートファジーに必要とされるタンパク質をコードしているのに対し、FUS/TLS(fused in sarcoma)およびTARDBP/TDP-43はともにRNA代謝において機能します(表4.7) *36,45 。Als2/Alsin遺伝子は若年発症、常染色体劣性型の家族性ALSに関与します(平均年齢6.5)*36,41,45。

病態生理学
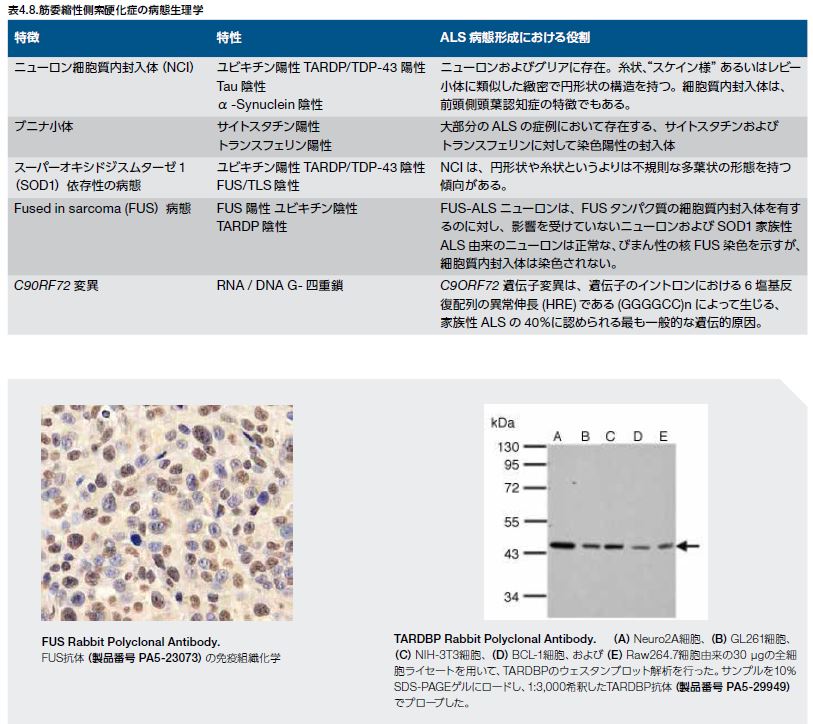
ALSの病態は、アストロサイトグリオーシス(ニューロン死に反応したアストロサイト増加)を伴う運動ニューロンの変性および死を特徴とします *48 。ユビキチン陽性、TARDBP陽性のニューロン細胞質内封入体(不溶性タンパク質凝集体)は、実質的に、SOD1またはFUS変異によるALSを除く全てのALS形態の顕著な特徴です *36, 38,49-51 。このため、疾患特有の病態生理学および/または疾患特有の遺伝子関連性に基づいて、ALSの様々な亜型を定義することができます。ニューロン細胞質内封入体は、それ自体が細胞死に寄与しているかは未知ですが、ALSの診断マーカーとされています *49,52 。ALSに関連する特性の概要を表4.8に示します。
ターゲットおよび有望な治療薬
既存のALSに対する薬物療法は、主に対症的で、疾患進行を遅延または回復させるよりも、痙攣、痙縮、唾液分泌過多、およびその他の症状の影響を軽減させることを目的としています。人工呼吸器も呼吸を補助する目的で使用されます。 ニューロン死からの保護や疾患進行の緩和を可能とする薬剤を発見するための努力が続けられていますが、いまだ成功にいたっていません。FDA承認薬の1つであるリルゾールは、わずかな生存期間延長効果(生存期間中央値が約3ヶ月延長)を示しますが、これはN-メチル-D-アスパラギン酸(NMDA)受容体活性と電位依存性ナトリウムチャネル活性の抑制および/またはグルタミン酸興奮毒性を減少させることによってもたらされていると考えられます *53 。
ALS亜型に特有の特徴を利用する新しい戦略がある一方、一般的な細胞プロセスや神経変性メカニズムをターゲットとするものもあります(表4.9) *54-62 。前者の例として、家族性SOD1の病態は特有で優性なSOD1含有封入体の毒性作用を示すため、変異タンパク質のレベルを減少させるアンチセンスオリゴヌクレオチド戦略は有益と考えられます。幹細胞療法は、運動ニューロン回路の修復あるいは神経栄養因子の供給、さらにはALS/FTD関連変異を有するALSおよび前頭側頭葉認知症(FTD)のヒトニューロンを用いるin vitroモデルの開発に有望な新しい方法です。治療において、これらのアプローチは、神経幹細胞、造血細胞、または人工多能性幹細胞由来神経前駆細胞のCNSへの移植が想定されます。
 目的のターゲットに特異的な抗体および免疫アッセイについてはこちらをご覧ください。
目的のターゲットに特異的な抗体および免疫アッセイについてはこちらをご覧ください。
多発性硬化症
多発性硬化症の特徴
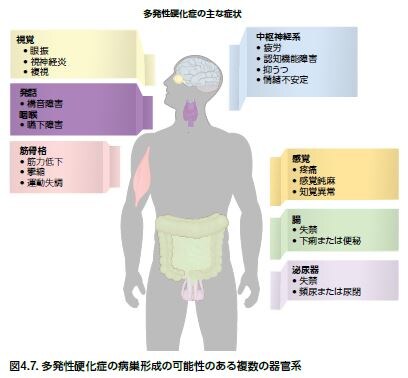
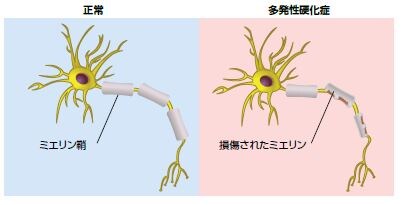
多発性硬化症(MS)は、脳および脊髄における慢性進行型の神経変性疾患で、炎症、脱髄、および軸索喪失を特徴とします *63-65 。MSには多くの形態があり、約90%の患者は少なくともある程度の回復および臨床的寛解と再発を繰り返します。二回目の継続的な疾患進行および増悪期に入った患者の50%以上において、通常、経時的に能力障害が生じます *63,66,67 。再発は、病巣の解剖学的部位、発作の発症、持続時間、および頻度、ならびに寛解の質によって著しく異なります *66 。MS発作の一般的な臨床症状は、四肢の麻痺または刺痛、筋力低下、平衡障害、視覚障害、および膀胱/腸の機能障害、ならびにその他の症状です(図4.7) *63 。MSに対する治療薬は存在しませんが、現在承認されている療法によって症状を軽減し、疾患進行を遅延できる可能性があります。

一般的な原因およびリスクファクター
約20%のMS患者には、MSの家族歴があり、MS発症に関連する遺伝要因が見られます *68 。例えば、同性の二卵性双生児におけるMSの一致率は5%であるのに対し、一卵性双生児におけるMSの一致率は約20~40%です *67,69-71 。主要組織適合性複合体(MHC)クラスII遺伝子HLA-DRB1は、MS発症と強い関連を持つ血清学的マーカーHLA-DR2およびHLADQ6に対応するハプロタイプの一部です *68,69 。これらのHLAタンパク質は、T細胞への抗原提示に関与する細胞表面抗原として働くHLAタンパク質です *68 。数多くの環境要因がさらにMS発症の非遺伝的リスクとして関与している可能性が提言されています。地理的条件とMS有病率は関係しており、日光への曝露量の低下に伴うビタミンD産生量の低下と、赤道からの距離が離れている地域での有病率の増加に一致が見られます。他のMS発病リスクを増加させると考えられる環境要因としては、喫煙や感染症罹患が考えられます *72 。
病態生理学
MSは、軸索損傷および脱髄を引き起こし、MSの発病および進行に寄与するCNS修復機構の障害と免疫調節不全を特徴とする複合疾患です(図4.8)*64,65 。MSの顕著な特徴は、白質および灰白質全体にわたるCNSの脱髄損傷や様々なサイズで不規則に散在するプラークです。これらは、脳室、視覚系、脳幹、および脊髄の近くで最もよく見られます *73 。ミエリン塩基性タンパク質(MBP)には、CNSを構成するタンパク質成分の約30%が含まれます。MBP(またはCNS関連ペプチド)をアジュバントとともに皮下投与することで、実験的アレルギー性脳脊髄炎(EAE)の十分に確立されたMSマウスモデルが誘導されます。また、MBPはMS患者の急性脱髄発症の脳脊髄液における検出を可能とするタンパク質バイオマーカーでもあります。

免疫病理学
MSにおける免疫応答の調節不全は、マクロファージ(MΦ)、樹状細胞(DC)などの抗原提示細胞(APC)による外来抗原および自己抗原のプロセシングおよび提示に関わります。APC表面のMHC分子に結合した同種抗原を認識し、続いて活性化末梢性T細胞が血液脳関門(BBB)を通過します。活性化B細胞もBBBを通過します。CNSにおいて、T細胞は、CNS関連抗原を提示するミクログリア細胞やアストロサイトにより再活性化されると、炎症誘発性サイトカインを分泌します。炎症誘発性のCNS微小環境におけるマクロファージは、腫瘍壊死因子α(TNF-α)を発現し、破壊的なスーパーオキシドおよび一酸化窒素種を放出します(図4.9)。
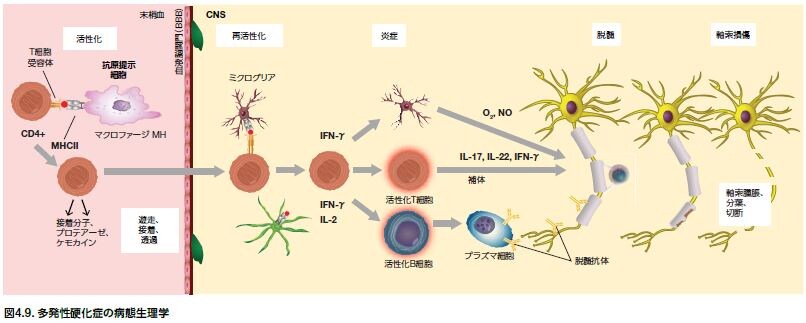
B細胞もまた抗原をT細胞に提示し、炎症性サイトカインを分泌し、マクロファージおよびミクログリア細胞を活性化する脱髄抗体を産生します。脱髄抗体によって駆動される補体カスケードの誘導により、膜侵襲複合体(MAC)が構築され、細胞膜に孔が形成されます。これにより、ミエリン膜が破壊されます。複数の要因がT細胞、マクロファージ、B細胞、およびミクログリアの異常な活性を介する脱髄に寄与します。MSにおいて、ミエリン形成オリゴデンドロサイト(OLG)の免疫介在性アポトーシスおよび壊死は、MSプラークにおける細胞喪失を引き起こします *74,75 。軸索損傷は、MS患者における臨床的障害の主な原因と考えられます。タンパク質の軸索輸送障害は、多くの神経変性疾患に共通する特徴です。アミロイド前駆体タンパク質(APP)に対し特異的なMS抗体および非リン酸化ニューロフィラメント重鎖(NFH)は、軸索損傷の有用なマーカーになり得ます *76 。

MSにおけるT細胞の役割
MSにおいて、軸索損傷は数多くのメカニズムによって起こる可能性があります。自己反応性B細胞および骨髄由来細胞からの損傷に加えて、軸索はCD4+サブセット由来の細胞およびCD8+細胞傷害性T細胞によっても攻撃される可能性があります *77-80 。MS患者では対照群に比して、より高頻度にミエリン抗原に対するT細胞反応性が確認されていることが数多くの試験で示されています。3種類の十分に特性決定されたミエリン抗原として、ミエリン塩基性タンパク質(MBP)、プロテオリピドタンパク質(PLP)、およびミエリンオリゴデンドロサイト糖タンパク質(MOG)があります。しかしながら、T細胞活性化はミエリン損傷を生じるミエリン抗原の遊離に続発して起こるのか、あるいはT細胞が一次損傷の原因であるのか、両方かについては明らかにされていません *81,82 。
活性化リンパ球により分泌されたサイトカインは、組織損傷を促進または修復を阻害することにより、グリア細胞およびニューロンを直接攻撃します。細胞ストレスおよび変性の下流経路は、サイトカインによる細胞死受容体の関与、活性酸素種の生成、ミトコンドリア機能不全、あるいはグルタミン酸またはカルシウムに対する興奮毒性によって媒介されます *83-85 。
歴史的に、Th1細胞はMSにおける炎症の主要なメディエーターと考えられていました。現在では、IL-17およびIL-22を分泌するCD4+ Th17細胞がMSの治療に極めて重要なターゲットであることが認識されています(図4.10) *82 。MS病態形成に関与する様々な受容体および可溶性因子の概略を図4.10に示します。その多くはMS治療のための薬剤ターゲットとなる可能性があります *80,82,86-95 。

既存の治療法および有望なターゲット
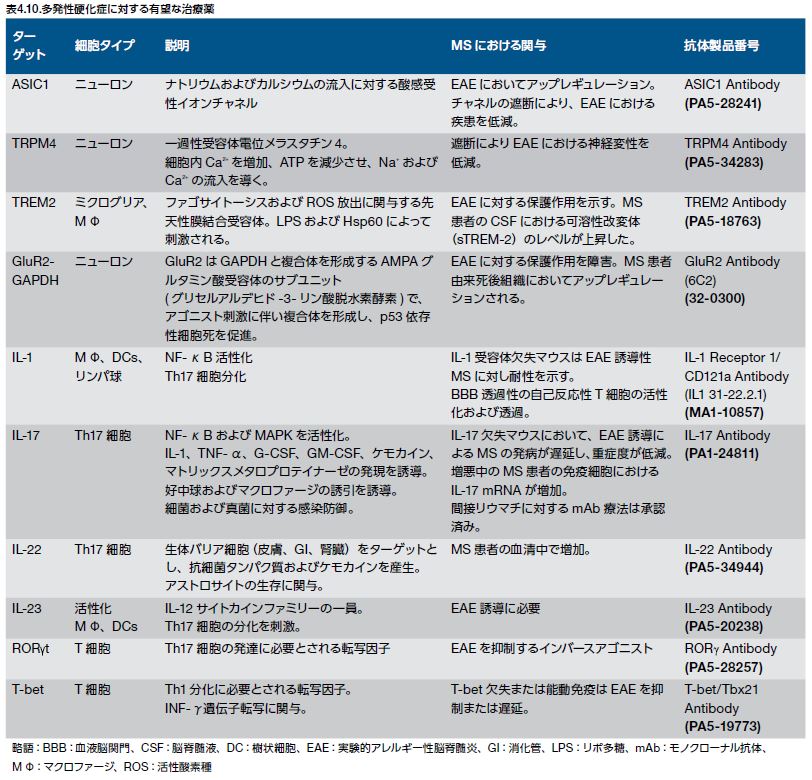
AD、PD、およびALSなどの他の神経変性疾患と比較して、MSに対しては数多くの疾患修飾療法が開発および承認されています *96 。
これらの薬剤には、インターフェロン、グラチラマー酢酸塩、生物製剤、および経口剤が含まれます(表4.11) *93,96-107 。
 既存の治療法に加えて、複数の疾患修飾治療薬がMSに対する有望な治療法として開発されています。現在、様々なアプローチが開発初期段階や後期段階にあります。免疫調節戦略には、自己T細胞療法、B細胞およびT細胞を介する免疫促進反応を抑制するモノクローナル抗体、T細胞受容体ペプチドワクチン、および他の新しいアプローチが含まれます。ようやく、間葉系幹細胞および人工多能性幹細胞の神経保護および/または神経変性特性に関する評価を行う臨床試験が進行中です *108-111 。
既存の治療法に加えて、複数の疾患修飾治療薬がMSに対する有望な治療法として開発されています。現在、様々なアプローチが開発初期段階や後期段階にあります。免疫調節戦略には、自己T細胞療法、B細胞およびT細胞を介する免疫促進反応を抑制するモノクローナル抗体、T細胞受容体ペプチドワクチン、および他の新しいアプローチが含まれます。ようやく、間葉系幹細胞および人工多能性幹細胞の神経保護および/または神経変性特性に関する評価を行う臨床試験が進行中です *108-111 。
当社では、MS発病に関わる研究プロセスで使用される一次抗体に加えて、サイトカイン定量用の高品質なELISAおよびLuminexアッセイも提供しています。詳しい情報についてはこちらをご覧ください。
【無料ダウンロード】神経変性疾患研究ガイドブック
当記事は、神経変性疾患研究ガイドブックからの抜粋です。以下の内容を含むPDFは下記から無料でダウンロード頂けます。
~主な内容~
- イントロダクション
- 神経変性疾患における共通のテーマ
- アルツハイマー病への取り組み
- 他の主要な神経変性疾患
- 抗体:神経変性疾患研究のための強力なツール
無料ガイドブックをダウンロードする
参考文献:
1. Poewe W (2006) The natural history of Parkinson’s disease. J Neurol 253 Suppl 7:VII2–VII6.
2. Farlow J, Pankratz ND, Wojcieszek J et al. (2004) Parkinson disease overview. [Updated 2014]. In: Pagon RA, Adam MP, Ardinger HH et al., editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2015. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1223/.
3. Kurtis MM, Martinez-Martin P (2013) Parkinson’s Disease: Symptoms, Unmet Needs and New Therapeutic Targets. Chapter 1 in: Martinex A, Gil C., editors, Emerging Drugs and Targets for Parkinson’s Disease. Cambridge, UK: The Royal Society of Chemistry. pp 3–25.
4. Klein C, Westenberger A (2012) Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med 2(1):a008888.
5. PINK1 Functions Medscape.com. http://www.medscape.com/viewarticle/711447_3. Accessed August 2015.
6. van Veen S, Sørensen DM, Holemans T et al. (2014) Cellular function and pathological role of ATP13A2 and related P-type transport ATPases in Parkinson’s disease and other neurological disorders. Front Mol Neurosci 7:48.
7. Murphy KE, Gysbers AM, Abbott SK et al. (2014) Reduced glucocerebrosidase is associated with increased -synuclein in sporadic Parkinson’s disease. Brain. 2014;137(Pt 3):834–848.
8. Lesage S, Brice A (2009) Parkinson’s disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet 18(R1):R48–R59.
9. Stefanis L (2012) -Synuclein in Parkinson’s disease. Cold Spring Harb Perspect Med 2(2):a009399.
10. Irwin DJ, Lee VM, Trojanowski JQ (2013) Parkinson’s disease dementia: convergence of -synuclein, tau and amyloid- pathologies. Nat Rev Neurosci 14(9):626–636.
11. Bendor JT, Logan TP, Edwards RH (2013) et al. The function of -synuclein.Neuron 79(6):1044–1066.
12. Lin X, Parisiadou L, Gu XL, Wang L et al. (2009) Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-diseaserelated mutant alpha-synuclein. Neuron 64(6):807–827.
13. Hebron ML, Lonskaya I, Moussa CE (2013) Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of -synuclein in Parkinson’s disease models. Hum Mol Genet 22(16):3315–3328.
14. Brundin P, Atkin G, Lamberts JT (2015) Basic science breaks through: new therapeutic advances in Parkinson’s disease. Mov Disord 30(11):1521–1527.
15. Stefanis L. -Synuclein in Parkinson’s disease. Cold Spring Harb Perspect Med. 2012 Feb;2(2):a009399.
16. Chandra S, Gallardo G, Fernández-Chacón R et al. (2005) Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 123(3):383–396.
17. Luk KC, Lee VM (2014) Modeling Lewy pathology propagation in Parkinson’s disease. Parkinsonism Relat Disord 20 Suppl 1:S85–S87.
18. Prescription Medications. Parkinson’s Disease Foundation. http://www.pdf.org/en/parkinson_prescription_meds. Accessed August 2015.
19. AlDakheel A, Kalia LV, Lang AE (2014) Pathogenesis-targeted, disease-modifying therapies in Parkinson disease. Neurotherapeutics 11(1):6–23.
20. Atala A, Lanza R, editors (2013) Introduction to Stem Cells. In: Handbook of Stem Cells. Volume 1 Pluripotent Stem Cells. Boston: Elsevier; 2013: 3.
21. Sandoe J, Eggan K (2013) Opportunities and challenges of pluripotent stem cell neurodegenerative disease models. Nat Neurosci 16(7):780–789.
22. Development of iPS From Donated Somatic Cells of Patients With Neurological Diseases. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT00874783?term=%22parkinson%27s%22+AND+%22stem+cell%22&rank=11. Accessed August 2015.
23. Paulson HL, Albin RL (2011) Huntington’s Disease: Clinical Features and Routes to Therapy. In: Lo DC, Hughes RE, editors. Neurobiology of Huntington’s Disease: Applications to Drug Discovery. Boca Raton (FL): CRC Press
24. WHO. Genes and human disease. http://www.who.int/genomics/public/geneticdiseases/en/index2.html#HD. Accessed August 2015.
25. Bates GP, Dorsey R, Gusella JF et al. (2015) Huntington disease. Nat Rev Dis Primers 11–21.
26. ClinicalTrials.gov. VX15/2503 Treatment for Huntington Disease (SIGNAL). https://clinicaltrials.gov/ct2/show/ NCT02481674?term=%22Huntington%22&recr=Open&no_unk=Y&rank=15. Accessed August 2015.
27. Warby SC, Graham RK, Hayden MR (1998) Huntington Disease. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle.
28. Prime View: HUNTINGTON DISEASE. Nature Reviews: Disease Primers. http://www.nature.com/public/article-assets/npg/nrdp/primeviews/huntingtondisease-2015.pdf, 2015. Accessed August 2015.
29. Bear MF, Connors BW, Paradiso MA (2007) Neuroscience: Exploring the Brain. 3rd ed. Philadelphia (PA): Lippincott Williams & Wilkins.
30. Papoutsi M, Labuschagne I, Tabrizi SJ et al. (2014) The cognitive burden in Huntington’s disease: pathology, phenotype, and mechanisms of compensation. Mov Disord 29(5):673–683.
31. Kumar A, Kumar Singh S, Kumar V et al. (2015) Huntington’s disease: an update of therapeutic strategies. Gene 556(2):91–97.
32. Zuccato C, Valenza M, Cattaneo E (2010) Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol Rev 90(3):905–981.
33. Pharm I. ISIS-HTTRx in a Phase 1/2a clinical study in patients with HD. https://www.isispharm.com/pipeline/.
34. Shannon KM, Fraint A (2015) Therapeutic advances in Huntington’s Disease. Mov Disord 30(11):1539–1546.
35. Zizkova M, Sucha R, Tyleckova J et al. (2015) Proteome-wide analysis of neural stem cell differentiation to facilitate transition to cell replacement therapies. Expert Rev Proteomics 12(1):83–95.
36. Renton AE, Chiò A, Traynor BJ (2014) State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 17(1):17–23.
37. Swinnen B, Robberecht W (2014) The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol 10(11):661–670.
38. Wijesekera LC, Leigh PN (2009) Amyotrophic lateral sclerosis. Orphanet J Rare Dis i4:1–22.
39. Hyperreflexia. TheFreeDictionary.com. http://medical-dictionary.thefreedictionary.com/hyperreflexia. Definition of hyperreflexia. Accessed August 2015.
40. Hypertonia. TheFreeDictionary.com. http://medical-dictionary.thefreedictionary. com/hypertonia. Accessed August 2015.
41. Kinsley L, Siddique T. Amyotrophic Lateral Sclerosis Overview. 2001 Mar 23 [Updated 2015 Feb 12]. In: Pagon RA, Adam MP, Ardinger HH et al., editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2015.
42. Muscle Twitching (muscle fasciculations). MedlinePlus. http://www.nlm.nih.gov/medlineplus/ency/article/003296.htm. Accessed August 2015.
43. Hyporeflexia. ALS Association. http://www.alsa.org/research/glossary/. Accessed August 2015.
44. Lillo P, Garcin B, Hornberger M et al. (2010) Neurobehavioral features in frontotemporal dementia with amyotrophic lateral sclerosis. Arch Neurol 67(7):826–830.
45. Bettencourt C, Houlden H (2015) Exome sequencing uncovers hidden pathways in familial and sporadic ALS. Nat Neurosci 8(5):611–613.
46. Turner BJ, Talbot K (2008) Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog Neurobiol 85(1):94–134.
47. Committee on the Review of the Scientific Literature on Amyotrophic Lateral Sclerosis in Veterans. Amyotrophic Lateral Sclerosis in Veterans: Review of the Scientific Literature. Washington DC: The National Academies Press; 2006.
48. Gliosis. TheFreeDictionary.com. http://medical-dictionary.thefreedictionary.com/gliosis. Accessed August 2015.
49. Mackenzie IR, Bigio EH, Ince PG et al. (2007) Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 61(5):427–434.
50. Vance C, Rogelj B, Hortobágyi T et al. (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323(5918):1208–1211.
51. Kwiatkowski TJ Jr, Bosco DA, Leclerc AL et al. (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323(5918):1205–1208.
52. Neumann M, Sampathu DM, Kwong LK et al. (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314(5796):130–133.
53. Wijesekera LC, Leigh PN (2009) Amyotrophic lateral sclerosis. Orphanet J Rare Dis 4:1–22,
54. The ALS Forum. New Findings on SOD1 Protein Properties Are Key Step in Antisense Clinical Trials—See more at: http://www.researchals.org/page/4832/14730/#sthash.p7SVrD6H.dpuf. http://www.researchals.org/page/4832/14730/. Accessed August 2015.
55. The ALS Association. New Award Will Lay Groundwork for C9orf72 Clinical Trials. http://www.alsa.org/news/archive/groundwork-for-c7orf72-trials.html. Accessed August 2015.
56. Cytokinetics. Press Releases. Cytokinetics and the ALS association announce awarding of grant for phase 3 clinical trial and biomarker research collaboration. http://www.cytokinetics.com/press_releases/release/pr_1436828630. Accessed August 2015.
57. NEALS. A prospective, multicenter, randomized, double-blind, placebocontrolled, parallel group, phase 2 study to compare the efficacy and safety of masitinib versus placebo in the treatment of patients suffering from Amyotrophic Lateral Sclerosis (ALS). http://www.alsconsortium.org/trial.php?id=109. Accessed August 2015.
58. ALSTDI. Masitinib Phase 3 Clinical Trial Update. http://www.alstdi.org/news/masitinib-phase-3-clinical-trial-update/. Accessed August 2015.
59. NEALS. Ibudilast (MN-166) in Subjects With Amyotrophic Lateral Sclerosis (ALS) (IBU-ALS-1201). http://www.alsconsortium.org/trial.php?id=121. Accessed August 2015.
60. NEALS. A Pilot Study of Inosine in ALS. http://www.alsconsortium.org/trial.php?id=122. Accessed August 2015.
61. NEALS. A Phase 2, Randomized, Double Blind, Placebo Controlled Multicenter Study to Evaluate Safety and Efficacy of Transplantation of Autologous Mesenchymal Stem Cells Secreting Neurotrophic Factors (MSC-NTF) in Patients With ALShttp://www.alsconsortium.org/trial.php?id=126. Accessed August 2015.
62. Neuralstem. Neuralstem Cell Therapy for ALS. http://www.neuralstem.com/celltherapy-for-als. Accessed August 2015.
63. Tullman MJ (2013) Overview of the epidemiology, diagnosis, and disease progression associated with multiple sclerosis. Am J Manag Care. 2013;19(2Suppl):S15–S20.
64. Van der Walt A, Butzkeuven H, Kolbe S et al. (2010) Neuroprotection in multiple sclerosis: a therapeutic challenge for the next decade. Pharmacol Ther 126(1):82–93.
65. Wilkins A, Scolding N (2008) Protecting axons in multiple sclerosis. Mult Scler 14(8):1013–1025.
66. Olek MJ (2005) Differential Diagnosis, Clinical Features, and Prognosis of Multiple Sclerosis. In: Olek MJ, editor. Multiple Sclerosis. Etiology, Diagnosis, and New Treatment Strategies. United States: Humana Press, Inc.pp 15–53.
67. Demetriou M (2005) Multiple Sclerosis, Genetics, and Autoimmunity. In: Olek MJ, ed. Multiple Sclerosis. Etiology, Diagnosis, and New Treatment Strategies. United States: Humana Press, Inc; pp 103–112.
68. Cree BAC (2014) Multiple sclerosis genetics. Hand Clin Neuro 122: 193–209.
69. Ontaneda D , Hyland M, Cohen JA (2012) Multiple sclerosis: new insights in pathogenesis and novel therapeutics. Annu Rev Med 63:389–404.
70. Campagnolo DI, Vollmer TL. Multiple Sclerosis (Updated September 22, 2010). eMedicine from WebMD. Available at: http://emedicine.medscape.com/article/310965-overview. Accessed March, 2011.
71. Oksenberg JR, Baranzimi SE, Hauser SL (2007) Biological concepts of multiple sclerosis pathogenesis and relationship to treatment. In: Cohen JA, Rudick RA, editors. Multiple Sclerosis Therapeutics, 3rd ed. United Kingdom: Informa UK Ltd. pp 23–44.
72. Simon KC, Schmidt H, Loud S et al. (2015) Risk factors for multiple sclerosis, neuromyelitis optica and transverse myelitis. Mult Scler 21(6):703–709.
73. Lucchinetti CF, Parisi J, Bruck W (2014) The pathology of multiple sclerosis. Neurol 23:77–105.
74. Nair A, Frederick TJ, Miller SD (2008) Astrocytes in multiple sclerosis: a product of their environment. Cell Mol Life Sci 65(17):2702–2720.
75. Neuhaus O, Archelos JJ, Hartung HP (2003) Immunomodulation in multiple sclerosis: from immunosuppression to neuroprotection. Trends in Pharmacol Sci 24(3):131–138.
76. Haines JD, Inglese M, Casaccia P (2011) Axonal damage in multiple sclerosis Mt Sinai J Med 78(2):231–243.
77. Trapp BD, Nave KA (2008) Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci 31:247–269.
78. Abbas AK, Lichtman AH, Pillai S (2005) Cellular and Molecular Immunology. 5th Edition. Philadelphia (PA): Elsevier Saunders.
79. Wilkins A, Scolding N (2008) Protecting axons in multiple sclerosis. Mult Scler 14(8);1013–1025.
80. Ellwardt E, Zipp F (2014) Molecular mechanisms linking neuroinflammation and neurodegeneration in MS. Exp Neurol 262 Pt A:8–17.
81. Rinker II JR, Naismith RT, Cross AH (2006) Multiple Sclerosis: An Autoimmune Disease of the Central Nervous System? In: Cook SD, editor. Handbook of Multiple Sclerosis, 4th ed. New York (NY): Taylor & Francis Group, LLC pp 95-112.
82. Fletcher JM, Lalor SJ, Sweeney CM et al. (2010) T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol 162(1):1–11.
83. Gilgun-Sherki Y, Melamed E, Offen D (2004) The role of oxidative stress in the pathogenesis of multiple sclerosis: the need for effective antioxidant therapy. J Neurol 251:261–268.
84. Stys PK (2005) General mechanisms of axonal damage and its prevention. J Neurol Sci 233(1–2):3–13.
85. Lassmann H (2014) Multiple sclerosis: lessons from molecular neuropathology. Exp Neurol 262 Pt A:2–7.
86. Yaghmoor F, Noorsaeed A, Alsaggaf S et al. (2014) The Role of TREM2 in Alzheimer’s disease and other neurological disorders. J Alzheimers Dis Parkinsonism 4(5)pii:160.
87. Piccio L, Buonsanti C, Cella M (2008) Identification of soluble TREM-2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain 131(Pt 11):3081–3091.
88. Zhai D, Lee FH, D’Souza C et al. (2015) Blocking GluR2-GAPDH ameliorates experimental autoimmune encephalomyelitis. Ann Clin Transl Neurol 2(4):388–400.
89. Sand IBK and Krieger S. Emerging Strategies for the Treatment of Multiple Sclerosis. Future Neurology. 2012;7(2):193-207. http://www.medscape.com/viewarticle/760849_5. Accessed August 2015.
90. Di Padova F, Ryffel B, Quesniaux V (2003). IL-17A Family, Receptors, Proinflammatory Effects, and Production. In: Quesniaux V, Ryffel B, Di Padova R (editors), IL-17, IL-22 and Their Producing Cells: Role in Inflammation and Autoimmunity. Second Edition. New York: Springer Basel pp 37–54.
91. Jin W, Dong C (2013) IL-17 cytokines in immunity and inflammation. Emerg Microbes Infect 2(9):e60.
92. Gaweco A, Palmer S, Shamilov R et al (2013) EAE disease prevention by a potent oral ROR t inverse agonist INV-17: a promising safe and efficacious MS treatment. J Neurol Sci 333:e362–e363.
93. Racke MK, Yang Y, Lovett-Racke AE (2014) Is T-bet a potential therapeutic target in multiple sclerosis? J Interferon Cytokine Res 34(8):623–632. 94. Sabat R, Ouyang W, Wolk K (2014) Therapeutic opportunities of the IL-22–IL-22R1 system. Nat Rev Drug Discov 13(1):21–38.
95. Perriard G, Mathias A, Enz L et al. (2015) Interleukin-22 is increased in multiple sclerosis patients and targets astrocytes. J Neuroinflammation 16;12:119.
96. Medications. National Multiple Sclerosis Society. http://www.nationalmssociety. org/Treating-MS/Medications. Accessed August 2015.
97. Copaxone. National Multiple Sclerosis Society. http://www.nationalmssociety.org/Treating-MS/Medications/Copaxone. Accessed August 2015.
98. Copaxone (Glatiramer Acetate) Injection. RXList.com. http://www.rxlist.com/copaxone-drug.htm. Accessed August 2015.
99. Kieseier BC (2011) The mechanism of action of interferon- in relapsing multiple sclerosis. CNS Drugs 25(6):491–502.
100. Aubagio. National Multiple Sclerosis Society. http://www.nationalmssociety.org/Treating-MS/Medications/Aubagio. Accessed August 2015.
101. Valentine WJ, Godwin VI, Osborne DA et al. (2011) FTY720 (Gilenya) phosphate selectivity of sphingosine-1-phosphate receptor subtype 1 (S1P1) G proteincoupled receptor requires motifs in intracellular loop 1 and transmembrane domain 2. J Biol Chem 286(35):30513–30525.
102. Gilenya. National Multiple Sclerosis Society. http://www.nationalmssociety.org/Treating-MS/Medications/Gilenya. Accessed August 2015.
103. Nguyen T, Nioi P, Pickett CB (2009) The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem 284(20):13291–13295.
104. Minagar A, Alexander JS, Sahrajan MA et al. (2010) Alemtuzumab and multiple sclerosis: therapeutic application. Expert Opin Biol Ther 10(3):421–429.
105. Hartung H-P et al. (2002) Mitoxantrone In Progressive Multiple Sclerosis: A Placebo-Controlled, Double-Blind, Randomised, Multicentre Trial. Lancet 360:2018–2025.
106. Tysabri. Proposed Mechanism of Action. https://www.tysabrihcp.com/en_us/home/clinical-overview/moa.html. Accessed August 2015.
107. Blight AR, Henney HR 3rd, Cohen R (2014) Development of dalfampridine, a novel pharmacologic approach for treating walking impairment in multiple sclerosis. Ann N Y Acad Sci 1329:33–44.
108. Mullard A (2011) Success of immunomodulators in MS shifts discovery focus to neuroprotection. Nat Rev Drug Discov 1;10(12):885–887.
109. Wingerchuk DM, Carter JL (2014) Multiple sclerosis: current and emerging disease-modifying therapies and treatment strategies. Mayo Clinic Proc 89(2):225–240.
110. Safety and Efficacy of Intravenous Autologous Mesenchymal Stem Cells for MS: a Phase 2 Proof of Concept Study (MESCAMS). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02239393. Accessed August 2015.
111. Xiao J, Yang R, Biswas S et al. (2015) Mesenchymal stem cells and induced pluripotent stem cells as therapies for multiple sclerosis. Int J Mol Sci 16(5):9283–9302.
研究用にのみ使用できます。診断目的およびその手続き上での使用はできません。
記事へのご意見・ご感想お待ちしています

【やってみた】熱変性の温度を下げた影響をリアルタイムPCRで確かめてみた
PCR反応における熱変性(denature)は、アニー�...
Read More
神経科学におけるフラグメント解析:複数の疑問を解決する柔軟なツール
脳は驚くべき器官です。さらに驚くべきこと...
Read More
リキッドバイオプシーを活用した肺がんと悪性脳腫瘍に対する研究事例のご紹介
リキッドバイオプシーは血液や尿などの体液...
Read More
細胞培養関連の学習リソースまとめ~トレーニング・セミナー・ハンドブックなど~
「細胞培養」はライフサイエンス研究におけ...
Read More