リアルタイムPCRにおける適正な定量法は実験の目的によって異なります。
- 絶対定量法によって、ターゲットの実際のコピー数が決定されますが、絶対定量法は最も煩雑で困難な定量方法です。この方法には周到な計画と高度に正確な検量線作成が必要です。絶対定量はウイルス価の決定に多く使用されます。
- 比較定量法においても綿密な実験計画が必要ですが、得られるデータは正確なコピー数ではなくサンプル間の相対値になります。遺伝子発現研究にはこの方法がよく利用されています。比較定量法には、ΔΔCt法と検量線法の2種類があります。
今回は、リアルタイムPCRの主な4つのデータ解析方法についてご紹介します。
▼もくじ [非表示]
絶対定量
絶対定量法は、研究対象であるターゲットの実際のコピー数を決定することが必要な研究者にとっては最適なリアルタイムPCR解析法です。絶対定量を行うためには、濃度既知のターゲットテンプレートから数桁の希釈系列を作成し、リアルタイムPCRによって増幅させ、各ターゲット濃度をPCR結果のCt 値に対してプロットすることにより検量線を作成する必要があります。未知試料のCt 値とこの検量線から、コピー数を決定します。
検量線の作成の概要
絶対定量で使用する検量線では、ターゲットのコピー数が既知である必要があります。このため、検量線の作成に先立って、テンプレートが正確に定量されることが要求されますが、これが検量線設定の最も重要な点となります。図1では検量線のセットアップについて示しています。ターゲットが試料中に正確に2 × 1011コピー存在するとします。試料は10倍希釈を8回段階希釈し、2 × 103コピーまで希釈します。各希釈液に対して少なくとも3 反復でリアルタイムPCRを実行します。得られる検量線によって、各コピー数とそれぞれのCt値との相関が得られます。この検量線との比較により、未知試料のコピー数が導き出されます。定量の精度は検量線の品質に直接関連しています。

図1 絶対定量向け検量線セットアップのためのワークフロー
絶対定量においては以下の点に留意してください。
-
- 検量線作成用のテンプレート調整、およびそのテンプレート量を正確に定量しておくことは、実験の基礎となります。また段階希釈におけるピペッティングの正確さは極めて重要です。リアルタイムPCRは非常に高感度であるため、些細な人為的エラーでも結果に影響してしまう場合もあるので注意してください。
- ターゲットテンプレートおよび実際の試料の段階希釈サンプルにおいて、RT効率及びPCR効率がほぼ一致することは非常に重要です。
検量線作成のためのテンプレートの選択
先述したように、絶対定量のための検量線作成にどのようなテンプレートを使用するかにより、データの正確さが決定されます。検量線作成用テンプレート中のコピー数の確認には、均一で純度の高い状態のものを利用されると思いますが、検量線作成時には、検査対象のサンプルの状態とできるだけ同レベルの純度のテンプレートを用いることが望ましいです。核酸の単離および逆転写などのステップで量の変化が起き得るため、ターゲットテンプレートは、実験試料が関与するのとほぼ同様の反応ステップに関与することとなります。以下のタイプのテンプレートが絶対定量のスタンダードとして使用されています。
- DNAスタンダード:興味対象ターゲットのPCRアンプリコンまたは興味対象ターゲットを含むプラスミドクローン。
- 利点:生成、定量が容易で、適切な保存により容易に安定性を維持できる。
- 欠点:リアルタイムPCRのRTの鋳型として使用できないため、RT効率の差を評価できない。
- RNA スタンダード:ターゲットのin vitro 転写RNA(図2)
- 利点:RT効率を評価でき、ターゲットの最も良いスタンダードとなる。
- 欠点:生成に時間がかかり、また不安定であるため長期保存が困難。
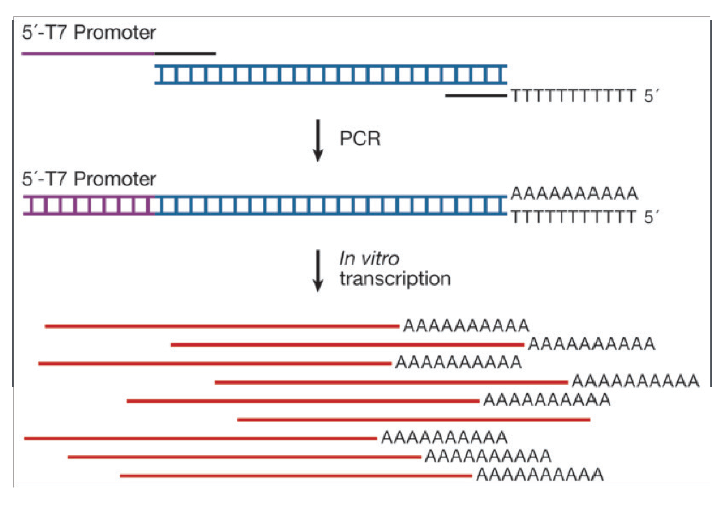
図2 in vitro 転写プロトコルの図解。リアルタイムPCRから生成されたPCR産物自身が5′ T7 プロモーターを含む配列および3′ poly(T)を含むリバースプライマーを用いて増幅されます。in vitro の転写反応によりポリアデニル化したセンスmRNAが生成されます。精製後、正確に定量し、検量線用に希釈します。
各RNAおよびDNAスタンダードは均質であるため、しばしば実験試料よりも反応効率が高くなります。より実験試料に近い不均質な環境を形成して反応効率のバランスをとるために、バックグラウンドRNAとして酵母tRNAなどをスタンダードテンプレートに添加することが可能です。バックグラウンドRNAにより、cDNA合成速度を10倍まで抑制可能であることが示されています。
検量線のアプリケーション – cRNA
絶対定量のために最適な検量線はどのように作成すればよいかを示すため、このセクションでは、絶対定量法のためのcRNA検量線の作成をステップ毎にご紹介していきます。
T7 RNAポリメラーゼは、プラスミドまたはPCR産物をテンプレートとして、対象遺伝子の転写産物のプールを作製するために使用することが可能です。
cRNAの測定には、UV吸収測定よりも検出限界の幅が広く精度がより高い蛍光測定が推奨されます。デジタルPCRを用いると、高精度で定量することができます。
コピー数を算出した後、検出に影響を与えない酵母tRNAをcRNAとtRNAの比が1:100となる割合で添加し、生体試料由来のバックグラウンドを再現することが可能です。この標準溶液を少なくとも5~6桁にわたって希釈し、リアルタイムPCRによるCt値の決定に使用します。
ピペッティングの不正確さは絶対定量のデータに極めて大きな影響を及ぼします。適切な注意を払うことによりこの影響を最小限に抑えることが可能です。ここに示すプレートセッティングでは、3種類の異なるcRNA段階希釈系列を作製し、それぞれの各希釈段階について2反復で増幅反応を行いました。
段階希釈溶液の濃度範囲は、実験に測定する試料中のターゲット遺伝子濃度を全てカバーすることが重要です。例えば、未知の試験試料に10コピーの遺伝子しか含まれない可能性がある場合、最小ポイントが100コピーのテンプレートまでの検量線では不十分です。
各希釈段階に関して、6個のCt 値が得られます。最大と最小のCt 値を除外して、残りの4個のCt 値の平均を算出します。例えば、この例で10–4 の希釈段階に注目すると(図3)、同一試料のCt値に2サイクルものばらつきがあることがわかります。このばらつきは、特定のコピー数に相当するこの希釈段階のCt 値を平均値の21.4とすることにより最小限に抑えられます。

図3 検量線作成のためのプレートセットアップ。サイクル閾値(Ct)の平均値を算出する場合には、反復反応から得られる、最大値および最小値は排除されます。
比較定量
比較定量も技術的には困難ですが、絶対定量ほど厳格ではありません。比較定量は多くの遺伝子発現研究に使用されている技術であり、キャリブレーター(ノーマル)試料および1つ以上の実験試料存在下で、興味対象遺伝子の発現量の変動を解析できます。この方法においては正確なコピー数決定は必要ではなく、キャリブレーター試料と比較した相対比に焦点を当てます。
ここでは、比較定量の一般的な方法の概要を示し、各方法におけるばらつきを最少限に抑える方法をご紹介します。
比較定量アルゴリズム – ΔCt
これは最も基本的な比較定量法です。興味対象遺伝子の発現に関して、試験試料およびキャリブレーター試料の両者からCt値が得られ、それらの差がΔCtとなります。相対比は単に2のΔCt 乗となります。
相対比 = 2ΔCt
この基本的な方法は、試料の量、品質、または反応効率の補正を行えないという点で、完全な方法とは言えません(図4)。
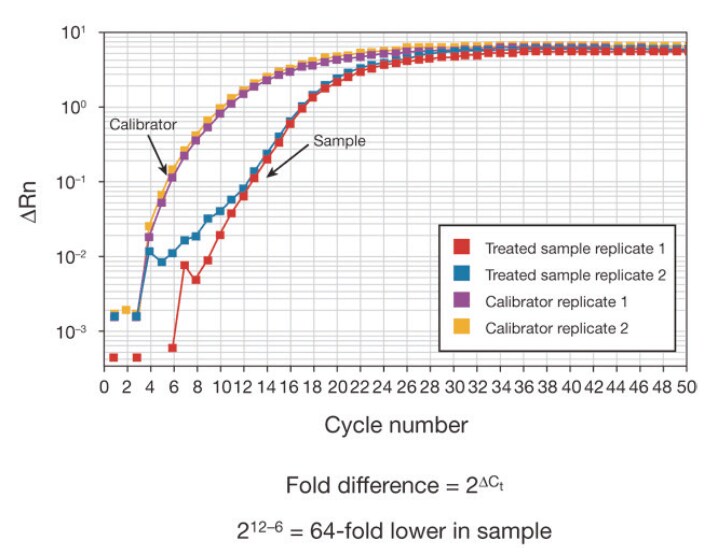
図4 処理サンプルとキャリブレーターにおける発現量の相対定量。処理サンプルおよびキャリブレーターを2反復でランしました。キャリブレーターのCt 値は6であり、処理サンプルのCt 値は12でした。ΔCt 法によると、処理サンプル中のターゲット遺伝子の相対発現量の計算値は、キャリブレーターより64倍低い値となりました。しかし、ノーマライザーを使用していないため、実験のばらつきがこの結果に及ぼしている影響が未知であり、結果は信頼性があるものとはみなされません。
比較定量アルゴリズム – ΔΔCt
ΔΔCt 法は非常に広く利用されている方法で、実験試料からの結果をキャリブレーター(例:未処理または野生型試料)およびノーマライザー(例:ハウスキーピング遺伝子発現)の両者の結果を比較します。この方法では、試験試料中およびキャリブレーター試料中の興味対象遺伝子(GOI)のCtが、同じ2つの試料中のノーマライザー(norm)遺伝子のCtに関して相対比較されています。得られるΔΔCt 値が発現相対比の決定に使用されます。
相対比 = 2-ΔΔCt
ΔCt試験試料 – ΔCtキャリブレーター = ΔΔCt
CtGOIS – CtnormS = ΔCt試験試料
CtGOIC – CtnormC = ΔCtキャリブレーター
ΔΔCt 法では、ノーマライザーおよびターゲット遺伝子の反応効率が同等であることが必要です。もちろん、誤差の許容範囲がどの程度であるかという疑問が生じるのは当然です。その決定には、同一試料を使用した、ノーマライザー遺伝子およびターゲット遺伝子両方の検量線を作成する方法が用いられます(図5)。各希釈サンプルに関して、ノーマライザーとターゲットの間の平均的ΔCtが得られます。重要なのは値そのものではなく、各希釈段階における値の一貫性です。
研究によっては、効率におけるこのわずかな誤差が不正確さの原因となる場合もあります。興味対象遺伝子およびノーマライザー遺伝子の両者に関する補正を行うことにより、増幅効率のばらつきによる効果を最小限に抑えることが可能です。
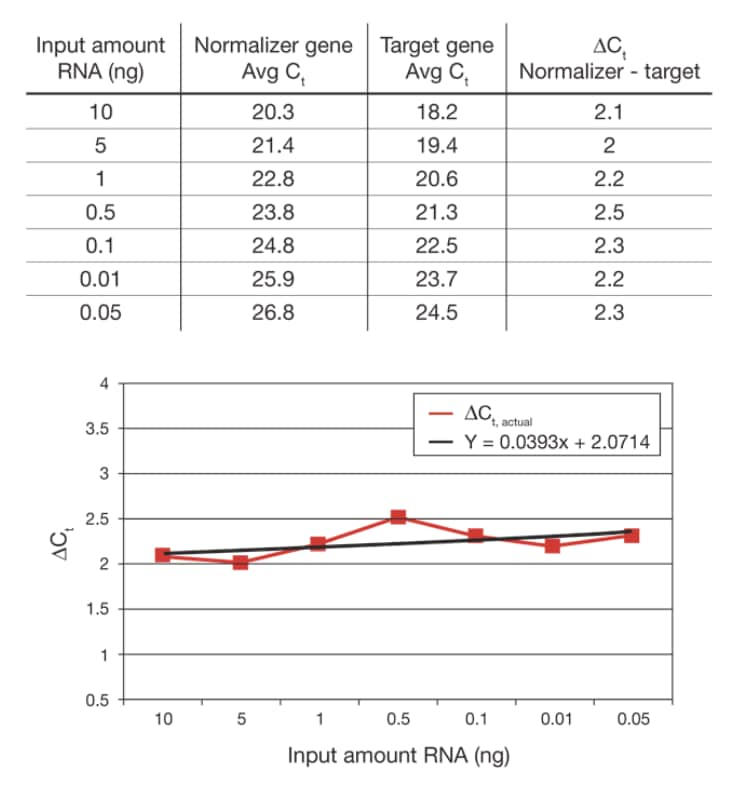
図5 相対効率プロット。ΔCt の範囲は2.0 ~ 2.5 です。ΔCtを希釈度またはRNAのインプット量に対してプロットし、傾きを求めます。完全に平らな曲線(傾き = 0)は、すべてのインプット濃度における効率が同等であることを示し、ΔΔCt 法においては、0.1 未満の傾きが許容範囲であると通常考えられています。
比較定量アルゴリズム – 検量線法
比較定量の検量線法は、試験試料中およびキャリブレーター試料中におけるターゲット遺伝子のCtの差を使用しますが、この値はレファレンス遺伝子CtSに対して標準化され、増幅効率のわずかなばらつきに対しても補正されています。この方法においては、ノーマライザーおよびターゲット遺伝子の両方の増幅効率を決定するための検量線が必要ですが、前述の方法で使用されていた仮定を排除することが可能です。この方法においては、ノーマライザー遺伝子が、解析対象の試料すべてにおいて同一であることが必要です。
Fold difference =(Etarget)ΔCttarget/(Enormalizer)ΔCtnormalizer
E = efficiency from standard curve E = 10[-1/slope]
ΔCttarget = CtGOIC – CtGOIS
ΔCtnormalizer = CtnormC– CtnormS
(M.W. Pfaffl 著 A-Z of Quantitative PCR から引用した相対比に関する等式。)
計算用に最も正確な相対値を得るためには、検量線のためのキャリブレーター試料を注意深く選択することが必要です。キャリブレーターは、実験試料と同じ精製過程で精製され、同じ反応条件で調整された、実験試料と同等の複雑性を有していることが必要です。このため、完璧なキャリブレーターは対象ターゲットを含む、不均質な試料であり、例えば研究中の細胞株または組織からのトータルRNAなどが含まれます。キャリブレーターと実験試料の差はすべて不正確な効率補正につながる可能性があり、その結果として遺伝子発現の相対比に関して不正確な結果が算出される可能性があるということに留意してください。コピー数は実測値ではないので、希釈または希釈値は任意な値にすることが可能です。
検量線法においても、ΔΔCt 法におけるのと同様の増幅曲線用の設定が使用されますが、ノーマライザーとターゲット遺伝子(GOI)のCt 値を調整するためには、検量線からの増幅効率値(理想的には同一のプレートでランされたもの)を使用します。効率は両者の傾きから導き出されます(図6)。
以上を要約すると、定量法の選択における第一段階は、まず絶対定量と相対定量のいずれが最適であるかを判断することです。試料中に存在するターゲット分子の数を知る必要がある場合には、既知量のターゲットテンプレートを使用して、正確な検量線を作成することが必要です。大部分の実験においては、相対的定量法が適切な方法となります。ΔCt法ではノーマライザーを使用しませんが、ΔΔCt 法ではリアルタイムPCRプロセスにおけるばらつきを補正するために1つ以上のレファレンス遺伝子を使用します。ノーマライザーを使用することにより、反応効率の調整を含む、あるいは含まない差の算出を行うことが可能です。
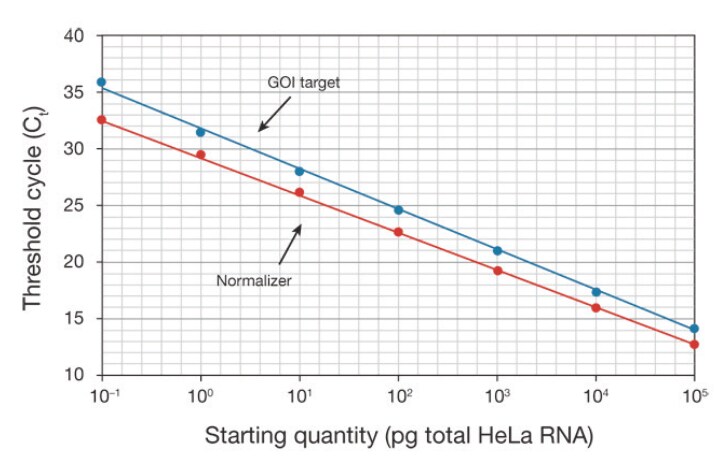
図6 ノーマライザーと興味対象遺伝子のCt 値を調整するために、検量線から増幅効率値が導き出されます。HeLa RNAを6オーダーの範囲で希釈し、リアルタイムPCRを行ってノーマライザーおよびターゲット遺伝子(GOI)両者に関する検量線を作成しました。
高分解能融解曲線(HRM)解析
高分解能融解曲線(HRM)解析は、SNP、新しい変異およびメチル化パターンを同定するための新しい方法です。この方法は同一条件でPCR後の密封チューブを用いて行います。HRM解析は従来の融解曲線法と比較してより感度の高い方法で、二本鎖のDNAが一本鎖のDNAに解離する温度をモニターします。この温度は、増幅産物の融点(Tm)として知られています。光学系および温度制御系の機器性能のアップグレードと共に、高速でのデータ回収と高精度の温度コントロールおよび均一性を実現するためのソフトウェアの解析機能を兼ね備えたリアルタイムPCR装置が必要です。QuantStudio 12K Flex、ViiA 7、7500 Fast、StepOnePlus およびStepOne Real-Time PCR SystemなどをHRM解析を行うことの可能な装置として現在販売しています。
HRM解析においては、約80~250塩基対の遺伝子フラグメントを高機能な二本鎖DNA(dsDNA)結合色素を含む反応液内でPCRにより増幅します。Applied Biosystems™ MeltDoctor™ HRM Master Mix では、二本鎖DNAの存在下、蛍光バックグラウンドが低く輝度の高いInvitrogen™ SYTO™ 9 色素の熱安定型であるMeltDoctor HRM Dyeを使用しています。40サイクルのランの後、増幅産物をアニールさせ蛍光度が最も高い状態にスタンバイさせます。HRM解析を開始すると、リアルタイムPCR装置はゆっくりと温度を上昇させると同時にアンプリコンからの蛍光データを記録します。PCR産物が変性(または融解)し始めると、蛍光色素が放出されるにつれて増幅産物の蛍光はそのTmに近づくまでゆっくりと低下していきます。Tmに最も近くなると、試料が二本鎖DNAから一本鎖DNAへと変化するのにあわせて蛍光の急激な低下が観察されます(図7)。
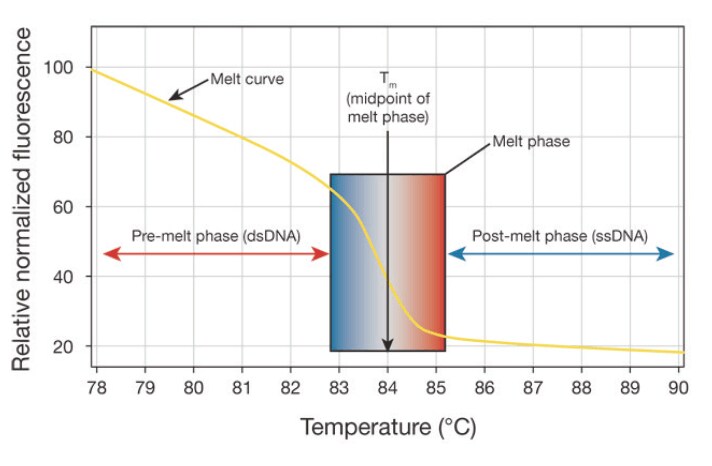
図7 PCRアンプリコンの融解曲線プロファイルの特性。個別のDNA配列毎に特徴的な融解曲線プロファイルが存在します。変異をTmのシフトまたは融解曲線の形の変化として検出することが可能です。従来の融解曲線解析とは対照的に、HRMでは単一ヌクレオチドの違いを有するアンプリコン間の識別が可能です。HRM法はdsDNA結合色素を用いた多くの新しいアプリケーションの可能性を広げます。
HRM解析アプリケーション
最も広く使用されているHRMアプリケーションは、遺伝子スキャニングです。遺伝子スキャニングは、PCRアンプリコンにおける未知の変異の検索で、シーケンシングに先立って、あるいはシーケンシングの代わりに行われます。PCR産物中の変異がHRMによって検出されるのは、変異があるとDNA融解曲線の形に変化が現れるためです。ヘテロ二量体のDNA試料は、増幅され融解すると、ホモ接合の野生型または変異型試料とは異なる融解曲線プロファイルを示します。
HRM解析の一般的なアプリケーションには以下のものが含まれます。
- 遺伝子スキャニング(変異検索)
- 変異解析
- 1塩基多型(SNP)検出
- ヘテロ接合性研究
- 種の同定
- メチル化解析
従来これらのアプリケーションには、各ターゲットに固有の蛍光プローブが必要でしたが、それらは高価で、デザインに時間がかかる上に柔軟性に欠けていました。dsDNA結合色素を使用するHRMは、プローブベースの解析と同様の性能を、より低価格で柔軟性のあるフォーマットを得ることができます。
HRMには多くのアプリケーションが存在しますが、Tm 値のわずかなシフトを検出する必要のある、単一塩基レベルでの識別が最も困難です。
HRM解析ケミストリ
特別な装置およびソフトウェアに加えて、HRM解析には単一ヌクレオチドの違いを有するアンプリコンの融点を識別する能力のあるdsDNA結合色素が必要です。HRMでの使用に成功している色素には、以下のものがあります。
- MeltDoctor™ HRM Dye
- SYTO™ 9色素
- LCGreen™ and LCGreen™ Plus+ reagents(Idaho Technology)
- EvaGreen™ dye( Biotium Inc.)
HRM解析アッセイ成功の鍵
- 最高の感度を達成するためには、アンプリコンを短くします。長いアンプリコンと比較して、100塩基対程度のアンプリコンでは単一ヌクレオチドの違いを有するアンプリコンの融点を識別することがより容易となります。
- PCRアンプリコンの特異性を確認します。ミスプライミング産物およびプライマーダイマーがあるとデータの解釈が複雑となります。高い特異性を得るためには、200nM未満のプライマー濃度を使用し、MgCl2 濃度を1.5mM~3mMの範囲とし、さらにホットスタートDNAポリメラーゼを使用することが有用です。標準的な低分解能の融解曲線を使用して、ミスプライミングの有無を評価してください。テンプレートを含まないコントロール(NTC)の融解曲線による特異性の評価を行うことが重要です。
- ターゲット以外の変異を含む領域での増幅を避けます。種相同性、エクソンとイントロンの境界、スプライス部位、およびPCR産物の二次構造および自己ヘアピン形成を確認します。
- 解析するすべてのターゲットに関して、蛍光プラトーが同様で、PCR産物の量が同様になるようにします。比較する試料間の量の差は、Tm値に影響を与え、HRM解析を混乱させる可能性があります。同量のテンプレートを使用することが有用な場合もあります。
- 反応に十分な量のテンプレートが使用可能なことを確認してください。一般的に、正確な融解解析に十分な産物を増幅するためにはCt値が30以下であることが必要です。
- 融解データの収集には十分な範囲の温度領域を設定してください。例えば、アンプリコンの融点の前後10°Cの範囲(Tm ± 10°C)の温度領域が必要です。正確な曲線の補正を行いかつ反復間での高い相関性を得るには、融点前後に十分なデータをとることが必要です。
- さらにいくつかの装置においては、すべての産物確実に再結合させて、ヘテロ二量体形成を促進するために、増幅に続いて(融解の前に)50°Cでのプレホールド段階を挿入することが推奨されます。ターゲット遺伝子フラグメントを高感度でバイアスなく増幅可能な、MeltDoctor HRM Master Mixなどのミックス試薬を使用してください。
マルチプレックスリアルタイムPCR解析
マルチプレックス反応は同一のリアルタイムPCR反応において複数のターゲットを解析する技術です。各ターゲットは、ターゲットに特異的な蛍光プローブまたはプライマーペアに結合した特定の色素により識別します。一般的に、マルチプレックス反応はノーマライザー遺伝子とターゲット遺伝子を同一の反応において増幅する目的で使用します。
理論的には、特定の反応中において増幅が可能なターゲット数は、使用できる光学的に識別可能な色素の数、およびリアルタイムPCR装置によって励起および検出が可能な色素の数によって制限されます。しかし、実際にはその他の実験的制約も存在します。例えば反応中において、異なるプライマーペアおよびプローブがお互いに反応しないことが必要であり、これらのプライマーおよびプローブはdNTPおよび熱安定性DNAポリメラーゼなどのPCR成分を共用しなければなりません。マルチプレックス反応は最適化に時間がかかりますが、いくつかの優れた利点が存在します。
- 変動が少なく、一貫性が高い。ノーマライザーとターゲット遺伝子を同一チューブ内でマルチプレックス反応することにより、これら2つのターゲットを別々の(隣り合っている場合でも)ウェルにおいて増幅することに起因する、ウェル間の誤差が排除されます。
- 使用する試薬の量が少なく、コストダウンにつながります。マルチプレックス反応では同数のターゲットの解析に必要な反応が少なくて済みます。
- より高いスループット。各PCRランおよび各試料についてより多くのターゲットの解析が可能となります。
マルチプレックスアッセイ成功の鍵
マルチプレックス反応を成功させるためには、以下に示す事項を含めた多くの要因を考慮する必要があります。
- プライマーおよびプローブのデザイン
- 試薬の最適化(プライマー濃度、ターゲットの量、反応成分および蛍光色素分子とクエンチャーの組み合わせなどを含みます)
- マルチプレックスアッセイの検証
プライマーおよびプローブのデザイン
プライマーおよびプローブのデザインは、マルチプレックスアッセイにおいて最も重要な因子です。反応の複雑性が増すにつれて、プライマーおよびプローブがダイマーとなる可能性、あるいは各反応成分の競合により複数のターゲットの増幅が制限される可能性が増加します。競合効果を最小限に抑え、性能を最大限に活かすために特に重要な因子を以下に示します。
- アンプリコンは短くします。50塩基対から150塩基対の範囲の部分を増幅するプライマーをデザインすることにより反応効率が上昇します。
- Tm値がお互いに2°C以内の範囲にあるプライマーをデザインします。すべてのプライマーおよびプローブが同一温度でアニールするということに留意してください。Tm値の違いは効率のバイアスにつながります。
- プライマーおよびプローブのデザインにはBLAST検索を行い、ターゲットに対する特異性を確認してください。
- プライマーデザインソフトウェアを使用して、プライマーまたはプローブにダイマー形成を起こしやすい配列がないかどうか確認してください。無料プログラムの一つである、AutoDimer(米国国立標準技術研究所のP.M.Vallone著)は特定のマルチプレックス反応におけるすべてのプライマーデザインに関して、すべての組み合わせにおけるダイマー形成の傾向を解析することができます。また、弊社のウェブサイトでMultiple Primer Analyzerをご利用いただくことができます。
試薬の最適化
マルチプレックス反応において極めて重要な問題は、増幅される異なるターゲット間での試薬の競合です。高い増幅効率を確保するためには、プライマーの量を減らすか、すべてのPCR成分の濃度を上昇させるか、あるいはその両方が必要です。一般的には、プライマー濃度の最適化から初めていただくケースが多いです。
プライマー濃度およびターゲットの量
マルチプレックス反応においてはすべてのアンプリコンが同数存在するわけではなく、また同じ効率で増幅するわけでもありません。より増幅しやすいあるいはより大量に存在するターゲットに対するプライマーの濃度を制限することにより、反応性を同レベルにすることが可能です。例えば、β-アクチン(高コピーノーマライザー)を存在量の少ないターゲットとマルチプレックスすると、β-アクチンがサイクルの初期の段階で、共用する反応成分を枯渇させてしまう可能性があります。β-アクチンプライマーの量を減少させることにより、増幅速度が制限され、より存在量の少ないターゲットを障害のない状態で増幅することが可能となります。一般的に、より存在量の多いターゲットに関しては、Ct 値を遅らせない範囲で最も低いプライマー濃度を使用することが必要です。
反応成分の濃度
個々のマルチプレックス反応毎に最適な反応条件は異なりますが、はじめにプライマー濃度を最適化することで、単独で反応したときと同程度のCt 値が得られる可能性が高くなります。その他の最適化項目として、Taq DNAポリメラーゼやマグネシウムおよびdNTPsの量を増やし、バッファー強度を高めることによって、反応系に含まれる全てのターゲットの感度および増幅効率を高めることができます。
蛍光色素分子とクエンチャーの組み合わせ
マルチプレックス反応におけるレポーター蛍光色素分子は、スペクトル的に特徴的で、各色素に由来する蛍光シグナルが単一チャネル内で検出されることが必要です。適切な色素を使用することにより、リアルタイムPCR装置の励起および光学系は、誤った蛍光色素に由来する蛍光の存在(このような障害はクロストークとも称されます)が発生しないように波長をフィルターすることが可能です。
同様に、マルチプレックスされるプローブの数が多くなると、各二重標識蛍光プローブにおけるクエンチャーの選択がより重要となってきます。TAMRA色素のような蛍光クエンチャーは、FAM色素用の一般的なクエンチャーであり、蛍光色素のエネルギーを異なる波長において放出することにより作用します。マルチプレックス反応においては、このタイプのクエンチャーは異なる波長における複数のシグナルを与え、フィルタリングが複雑化し、データの安定性にも影響する可能性があります。一方、ダーククエンチャーはエネルギーを蛍光ではなく熱の形で放出するため、全体的な蛍光バックグラウンドがより低く抑えられます。ご使用になられる装置の検出能に基づいて、適切なマルチプレックス色素の組み合わせを選択してください。多くの色素のスペクトル互換性に関しては、弊社の蛍光スペクトルビューアーをご覧ください。
マルチプレックスアッセイの検証
シングルプレックスのリアルタイムPCRの場合と同様に、アッセイのランの前に、マルチプレックス反応中のすべてのターゲットの反応効率を評価するために検量線を作成する必要があります。この検証過程は大きく2つの段階に分けられます。
- 各ターゲットのプライマーおよびプローブを検証し、個々の効率を同定します。これがプライマーおよびプローブデザインの初期の評価です。
- マルチプレックスアッセイ全体の最適化。
ターゲットにとって理想的なデザインおよび条件を決定するために、個々のターゲットに関するプライマーおよびプローブセットを評価してください。段階希釈の検量線を用いて評価します。100%に近い効率が得られることを目標としてください。各プライマーおよびプローブの組み合わせを機能的に評価した後、マルチプレックスの最適化に進んでください。プライマーおよびプローブの組み合わせに関するシングレックスの条件は、ターゲットがマルチプレックスである場合には最適とならない可能性があることに留意してください。マルチプレックスパネルを構築する際には、初回の実験においてすべてのターゲットを組み合わせるのではなく、1回に1個ずつターゲットを添加していくことが賢明です。
同様の検量線法を使用して、すべてのプライマーとプローブを混合し、各希釈段階に関してマルチプレックス反応を実行してください。各ターゲットに関して得られた効率を、対応するシングレックスのリアルタイムPCR反応の効率と比較してください。理想的には、シングル反応とマルチプレックス反応の間にはほとんど変化がないはずです。もしマルチプレックスターゲットの反応効率の変化が5%を超えているかあるいは好ましい範囲である90%~110%に収まらない場合には、プライマーまたはプローブの濃度またはマルチプレックスアッセイ中の他の成分の濃度を最適化する必要があります。
感度の低下を防ぐためには、マルチプレックス反応におけるCt 値がシングレックス反応において得られたCt 値と同等であることが必要です。
リアルタイムPCRハンドブック 無料ダウンロード
このハンドブックでは、リアルタイムPCRの理論や実験デザインの設計など、リアルタイムPCRの基礎知識が掲載されています。リアルタイムPCRを始めたばかりの方やこれから実験を考えている方にうってつけのハンドブックです。PDFファイルのダウンロードをご希望の方は、下記ボタンよりお申し込みください。
【無料公開中】リアルタイムPCRトラブルシューティング
異常なS字状の増幅曲線、NTCでの増幅検出、増幅が見られないなど、リアルタイムPCRの実験中に困った際の手助けになる情報を紹介しています。PDFファイルのダウンロードをご希望の方は、下記ボタンよりお申し込みください。
研究用にのみ使用できます。診断用には使用いただけません。
記事へのご意見・ご感想お待ちしています

【やってみた】熱変性の温度を下げた影響をリアルタイムPCRで確かめてみた
PCR反応における熱変性(denature)は、アニー�...
Read More
「PCRの達人」に聞く!デジタルPCRの魅力とは?
デジタルPCRは検量線サンプルを必要とせずに...
Read More
公共用水の安全を守る:デジタルPCRによる寄生虫モニタリング
Absolute Gene-iusは、先進的な研究者にインタビ�...
Read More
【やってみた】実験中に放置したままのゲノムDNAは沈むのか?リアルタイムPCRで確かめてみた
PCRやリアルタイムPCRでは、サンプル由来のDNA...
Read More